


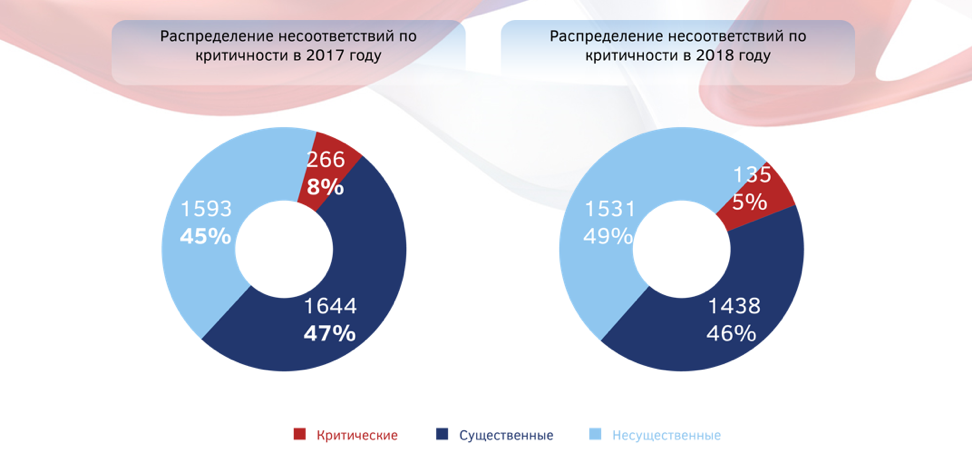
Какова процедура инспектирования иностранных производителей на соответствие правилам надлежащей производственной практики, какие несоответствия чаще всего встречаются на площадках, последствия для производителя, не устранившего замечания инспектората, - вот лишь небольшой перечень вопросов, которые волнуют фармсообщество и регулярно задаются инспекторату. Не стала исключением и конференция, посвященная стандартам GxP. По данным инспектората ФБУ «ГИЛС и НП» в 2018 году было выявлено 3 503 несоответствия, из которых 135 (5%)были классифицированы как критические, 1 438 - (46%)как существенные и 1 531 (49%)- как несущественные.
Если эти данные проанализировать через призму несоответствий конкретным главам правил GMP, закрепленных в Приказе Минпромторга России № 916 «Правила организации производства и контроля качества лекарственных средств», становится видно, что наибольшее количество несоответствий, влияющих на качество лекарственных препаратов и, как следствие, на жизнь и здоровье пациента, касается Приложения 1 и Главы 1:
Более подробно об особенностях и лучших практиках инспектирования фармпроизводств на соответствие стандартам GMP, гармонизации правил проведения инспекций в рамках единого рынка ЕАЭС, подходах к формированию корректирующих мероприятий по результатам выявленных в ходе инспекции несоответствийэксперты российского государственного GMP-инспектората и представители иностранных регуляторов будут говорить на двухдневном мастер-классе «GMP-инспектирование» в рамках GMP-конференции, которая пройдёт 23-25 сентября в Калининграде. |
Типичные несоответствия иностранных фармплощадок по главам правил GMP



1.1
2025CiteScore
43-й процентиль
Powered by 
































_2.jpg")
Популярные статьи
Том 8, № 2 (2019)
Том 8, № 3 (2019)

Уведомления
Облако тегов
ВЭЖХ
ВЭЖХ-МС/МС
ВЭЖХ-УФ
УФ-спектрофотометрия
биоэквивалентность
валидация
вспомогательные вещества
высокоэффективная жидкостная хроматография
доклинические исследования
количественное определение
контроль качества
лекарственное растительное сырье
острая токсичность
плазма
растворимость
стандартизация
таблетки
тест «Растворение»
фармакокинетика
фармацевтическая разработка
флавоноиды

117149, Москва, Симферопольский бульвар, 8
ООО «ЦФА»
e-mail: info@pharmjournal.ru
Свидетельство о регистрации СМИ ЭЛ № ФС 77 - 75437 от 01.04.2019 выдано Федеральной службой по надзору в сфере связи, информационных технологий и массовых коммуникаций
Обработка персональных данных