Научно-производственный рецензируемый журнал «Разработка и регистрация лекарственных средств» - актуальное бесплатное прикладное издание и информационный портал для специалистов, задействованных в сфере обращения лекарственных средств. Журнал предназначен для фармацевтических предприятий-производителей и их сотрудников из отделов разработки, контроля качества, регистрации, производства и развития; сотрудников лабораторных центров, контрактно-исследовательских организаций, научных и образовательных учреждений. Индексируется в российских и международных реферативных и полнотекстовых базах, включен в наукометрические базы данных РИНЦ и Scopus, входит в "Белый список" научных изданий и Перечень ВАК (категория К1) и Russian Science Citation Index (RSCI).
Наименование и содержание научных работ, публикуемых в журнале «Разработка и регистрация лекарственных средств», должно соответствовать отраслям науки:
- 3.3.6. – фармакология, клиническая фармакология (медицинские науки, фармацевтические науки, биологические науки);
- 3.4.1. – промышленная фармация и технология получения лекарств (фармацевтические науки);
- 3.4.2. – фармацевтическая химия, фармакогнозия (фармацевтические науки).
Основные пять тематических разделов журнала «Разработка и регистрация лекарственных средств» включают цикл развития лекарственного средства от его создания до получения регистрационного удостоверения. Первый раздел посвящен поиску и разработке новых лекарственных средств, второй - фармацевтической технологии и рассматривает научные и практические направления от разработки и производства исходных фармацевтических ингредиентов, технологий и оборудования – до создания стандартных и терапевтически эффективных лекарственных препаратов. Третий раздел описывает аналитические методики контроля качества; четвертый раздел посвящен подходам к оценке эффективности и безопасности лекарственных средств, проведению долклинических и клинических исследований; в пятом разделе рассматриваются вопросы валидации методик, подготовки регистрационного досье, жизненный цикл лекарственного препарата в GxP окружении.


Текущий выпуск

ОТ РЕДАКЦИИ

17–18 февраля 2026 года в Москве состоится ежегодный двухдневный конгресс «Разработка и регистрация лекарственных средств». Тематика конгресса традиционно охватывает все ключевые этапы жизненного цикла лекарственного средства, от разработки до пострегистрационных исследований.

В рамках X Всероссийской GMP-конференции прошла сессия, посвященная вопросам регуляторной науки в сфере высокотехнологичных лекарственных препаратов в Российской Федерации.

Открытая дискуссия, посвященная вопросам локализации препаратов CAR-T клеточной терапии в Российской Федерации, состоялась с участием представителей Государственной думы, федеральных органов исполнительной власти, экспертного сообщества, фармацевтической индустрии и ведущих медицинских центров 6–7 октября в рамках «БИОПРОМ: промышленность и технологии для человека».

В новом интервью из цикла «Мнение лидеров» мы поговорили с коммерческим директором компании «АИКС ЛАБ» Вербицкой Марией Борисовной, главным научным консультантом Electrolab India Pvt Ltd. доктором Параг Гадкари и генеральным директором Центра Фармацевтической Аналитики Игорем Шохиным.

В статье приведены факты о состоянии витаминной промышленности СССР в предвоенные годы. Перечислены научно-исследовательские и производственные организации, участвовавшие в исследовании и производстве витаминной продукции. Представлены данные экспериментов по работе с лекарственным растительным сырьем и стабилизации водных растворов с витамином С. Приведен способ очистки растворов аскорбиновой кислоты, полученной из хвои. Изучено содержание витамина С в различных видах лекарственного растения рода шиповник в процессе созревания. Описаны способы обогащения рыбьего жира витамином А из печени судака и экстрагирование каротина из отходов производства аскорбиновой кислоты. Представлена история обсуждения и утверждения отечественного витамина К на заседании Фармакологического комитета НКЗдрава СССР.

Компания «ФАРМ-АКТИВ ПРО» обеспечивает системный подход к оснащению фармацевтических и лабораторных производств. В нашем понимании поставка – это полный цикл, от проектирования и выбора оптимальных решений до ввода в эксплуатацию, квалификации и сопровождения в течение всего жизненного цикла. Все – с учетом требований регистрации лекарственных средств, валидности процессов, контроля качества и регуляторной совместимости.
МЕРОПРИЯТИЯ

19 сентября 2025 года в технопарке «Калибр» состоялось знаковое событие для специалистов фармацевтической отрасли — мастер-класс, посвященный актуальным аспектам разработки и регистрации лекарственных средств. В фокусе мероприятия – мастер-классы от экспертов «Центра Фармацевтической Аналитики», ориентированные на вопросы проведения регистрационных лабораторных исследований.
ПОИСК И РАЗРАБОТКА НОВЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ
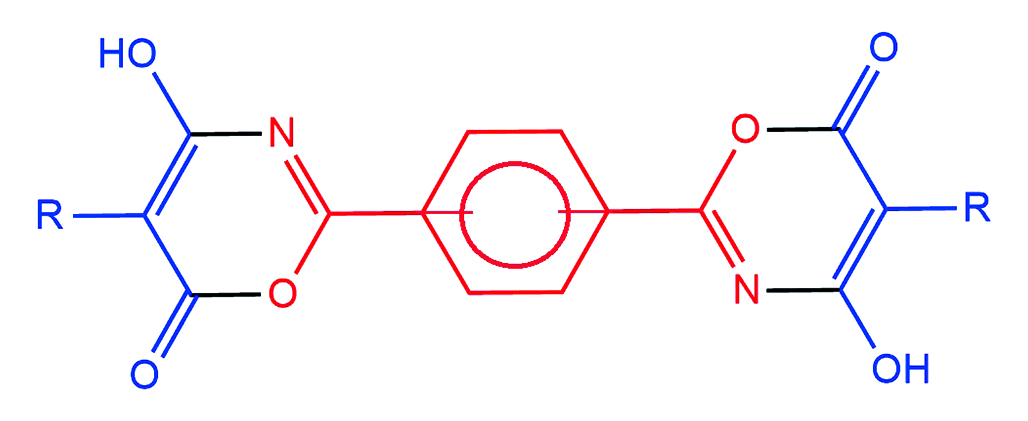
Введение. Исследование производных 1,3-оксазина играет ключевую роль в области химии гетероциклических соединений. Тем не менее в литературе отсутствует систематизированная информация о способах синтеза биспроизводных 1,3-оксазин-6-онов. Создание эффективных методов получения этих оксазинов, а также изучение их структуры, характеристик и биологической активности представляет собой многообещающее направление как для медицинской химии, так и для фармацевтической промышленности.
Цель. Разработать лабораторный способ получения бис(4-гидрокси-6H-1,3-оксазин-6-онов) на основе реакции бензол-1,3-дикарбоксамида и бензол-1,4-дикарбоксамида с замещенными малонилхлоридами и доказать их строение с помощью спектроскопии ядерного магнитного резонанса (ЯМР) на ядрах 1Н и 13С.
Материалы и методы. Спектры 1H- и 13C-ЯМР были зарегистрированы на приборе Bruker AM-600 в дейтерированном диметилсульфоксиде (ДМСО-d6), их обработка осуществлена с помощью программного обеспечения ACD/Labs. Мониторинг протекания реакций проведен с применением тонкослойной хроматографии на пластинах с силикагелем TLC Silicagel 60 F254 с использованием этилацетата в качестве элюента и детекцией в ультрафиолетовом (УФ) свете. Методика синтеза включала суспендирование 1 ммоль бензол-1,4-дикарбоксамда или бензол-1,3-дикарбоксамида в абсолютном 1,2-дихлорэтане, добавление 2,4 ммоль замещенного малонилхлорида и кипячение смеси в течение 15 ч. Конец реакции определен по отсутствию исходных диамидов в реакционной массе методом тонкослойной хроматографии.
Результаты и обсуждение. В результате исследования был разработан лабораторный метод синтеза бис(4-гидрокси-6H-1,3-оксазин-6-онов) с фениленовыми мостиками между гетероциклическими фрагментами. Использование в качестве растворителя абсолютного 1,2-дихлорэтана позволило увеличить выход целевых соединений на 5–7 % и сократить время синтеза на 5 ч по сравнению с абсолютным бензолом. Строение синтезированных веществ было доказано спектроскопией ядерного магнитного резонанса на ядрах 1H и 13C. В спектрах полученных соединений присутствуют все характеристичные сигналы, соответствующие бис(4-гидрокси-6Н-1,3-оксазин-6-онам).
Заключение. Разработан, реализован и оптимизирован лабораторный метод синтеза бис(4-гидрокси-6Н-1,3-оксазин-6-онов) на основе реакции бензол-1,3-дикарбоксамида и бензол-1,4-дикарбоксамида с монозамещенными малонилхлоридами. Целевые соединения были получены в абсолютном 1,2-дихлорэтане с выходом, близким к количественному. Это подтверждает эффективность разработанного подхода. Структура всех полученных бис(4-гидрокси-6Н-1,3-оксазин-6-онов) была достоверно установлена с помощью спектроскопии ядерного магнитного резонанса на ядрах 1H и 13C.
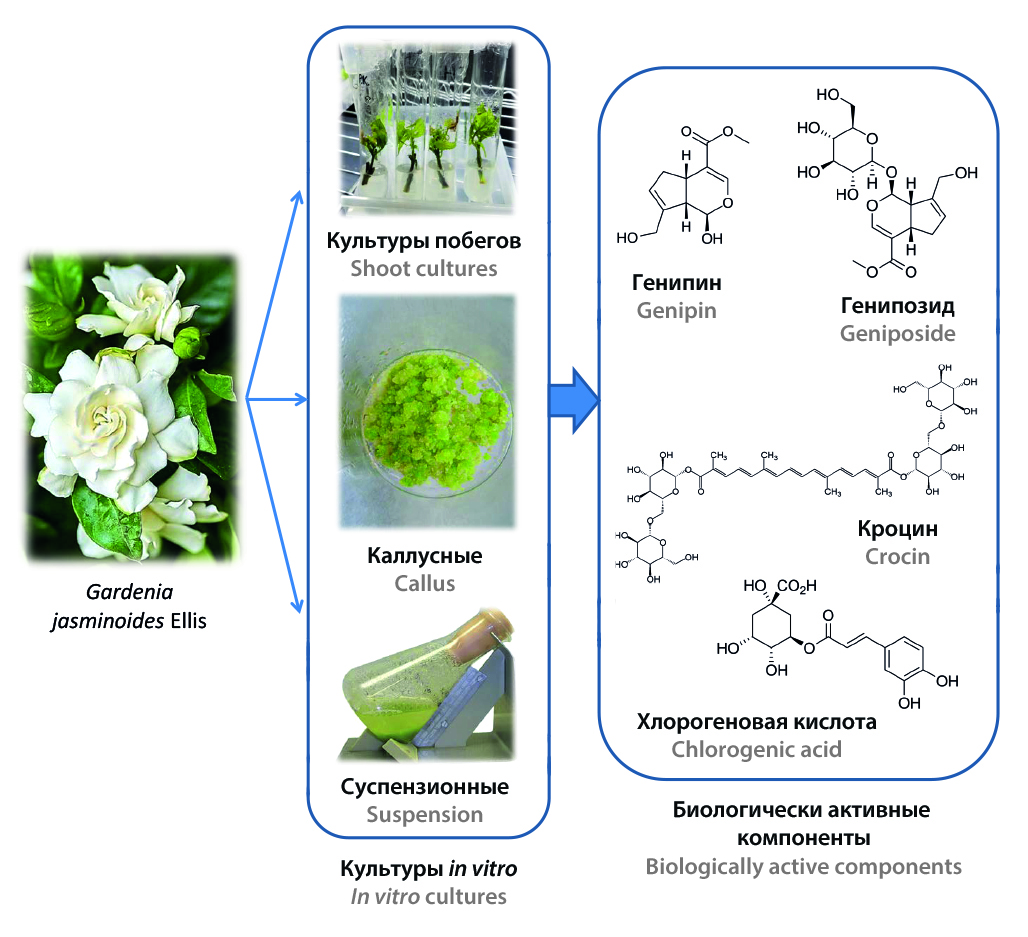
Введение. Гардения жасминовидная (Gardenia jasminoides Ellis) — вечнозеленый кустарник из семейства Rubiaceae, естественно произрастающий в Китае и Японии и активно культивируемый в качестве декоративного растения во многих регионах мира. В данном обзоре приведены данные о результатах исследований накопления биологически активных веществ (БАВ) в различных культурах, возможности индукции их биосинтеза и биологической активности БАВ, выделенных из культур гардении жасминовидной.
Текст. В настоящее время из интактного растения G. jasminoides выделено и идентифицировано около 162 соединений, среди которых флавоноиды, иридоидные гликозиды, желтый пигмент гардении, монотерпеноиды, сесквитерпеноиды, тритерпеноиды, органические кислоты и их производные. Наиболее важными биологически активными компонентами G. jasminoides являются иридоидные гликозиды и желтые пигменты – производные кроцина. Использование культур растительных клеток является одним из наиболее перспективных подходов к получению БАВ растительного происхождения, поскольку данный метод характеризуется меньшей зависимостью от климатических и экологических факторов, обеспечивает более точное управление процессом и позволяет сокращать производственный цикл, что способствует эффективному масштабированию производства. Рядом исследователей были получены каллусные и суспензионные культуры, а также культуры побегов и модифицированных корней G. jasminoides. С целью повышения продукции основных классов биологически активных веществ – иридоидных гликозидов, полифенольных соединений и каротиноидов – в каллусных, суспензионных и побеговых культурах гардении жасминовидной был проведен ряд исследований по введению в питательные среды специфических добавок. Было показано, что культуры клеток G. jasminoides обладают высокой антиоксидантной активностью благодаря фенольным соединениям, таким как феруловая и хлорогеновая кислоты. Экстракты каллусной культуры показали значительно большую супероксиддисмутазную активность, чем экстракты листьев. В то же время только экстракты каллусных культур проявляли антимикробную активность против Escherichia coli и Bacillus cereus.
Заключение. Обзор литературных данных позволяет заключить, что культуры G. jasminoides in vitro обеспечивают стабильное и усиленное производство ценных вторичных метаболитов (иридоидных гликозидов, полифенолов, каротиноидов), превосходящее показатели интактных растений, что открывает перспективы промышленного получения БАВ и препаратов на их основе.

Введение. В последние годы возросла актуальность использования лекарственных растений в составе лечебно-профилактических препаратов, способных регулировать адаптивные модификации, направленные на поддержание психических и физических функций человека в условиях постоянно изменяющихся природных и социальных факторов. Одним из интересных растений в лекарственном отношении является родиола розовая (Rhodiola rosea L.) – представитель семейства толстянковых (Crassulaceae). Данный вид является очень полиморфным как морфологически, так и с точки зрения химического состава. Основной сырьевой базой являются горы Южной Сибири (Алтай, Саяны), Тува, Забайкалье. Но отечественных сырьевых баз недостаточно, поэтому актуальна проблема их расширения и выявления среди культивируемых популяций фенотипов с высоким содержанием биологически активных веществ.
Цель. Установить морфометрические характеристики различных фенотипов родиолы розовой, культивируемой на северо-западе России и провести сравнительный анализ вторичных метаболитов в образцах подземных органов родиолы розовой разных фенотипов с использованием высокоэффективной тонкослойной хроматографии (ВЭТСХ).
Материалы и методы. Объектами исследования были 24 образца корневищ и корней родиолы розовой разных фенотипов, заготовленных в 2024 г. в фазы цветения – плодоношения в питомнике лекарственных растений СПХФУ (Ленинградская обл., Всеволожский р-н, 38 км Приозерского шоссе). Извлечения из сырья получали, используя ультразвуковую ванну «Сапфир-4,0-ТТЦ» (Россия). В качестве экстрагента использовали спирт этиловый 70%-й. ВЭТСХ-анализ выполняли на приборе CAMAG (Швейцария) c использованием пластин MERCK HPTLC plates silica gel 60 F 254 размером 20 × 10 см.
Результаты и обсуждение. Полученные растворы изучали методом ВЭТСХ в системе растворителей хлороформ – спирт 96%-й – вода (25 : 16 : 1). После проведения сканирующей денситометрии при 254 нм сравнивали денситограммы отдельных треков между собой для того, чтобы выявить фенотипы родиолы розовой с большим содержанием биологически активных соединений. В результате сравнения треков 24 образцов подземных органов Rhodiola rosea в числе перспективных для последующего исследования и культивирования предложены образцы женского (№ 1, 7, 8 и 14) и мужского (№ 15, 18, 21, 23 и 24) пола высотой от 23 до 41 см.
Заключение. Были установлены морфометрические характеристики различных фенотипов родиолы розовой, культивируемой в условиях северо-запада России. В результате анализа спиртовых извлечений из корневищ и корней Rhodiola rosea методом ВЭТСХ было установлено, что большим содержанием вторичных метаболитов (фенилпропаноидов и простых фенолов) отличаются образцы как женского, так и мужского пола. Высота растений варьировала от 23 до 41 см.
ФАРМАЦЕВТИЧЕСКАЯ ТЕХНОЛОГИЯ
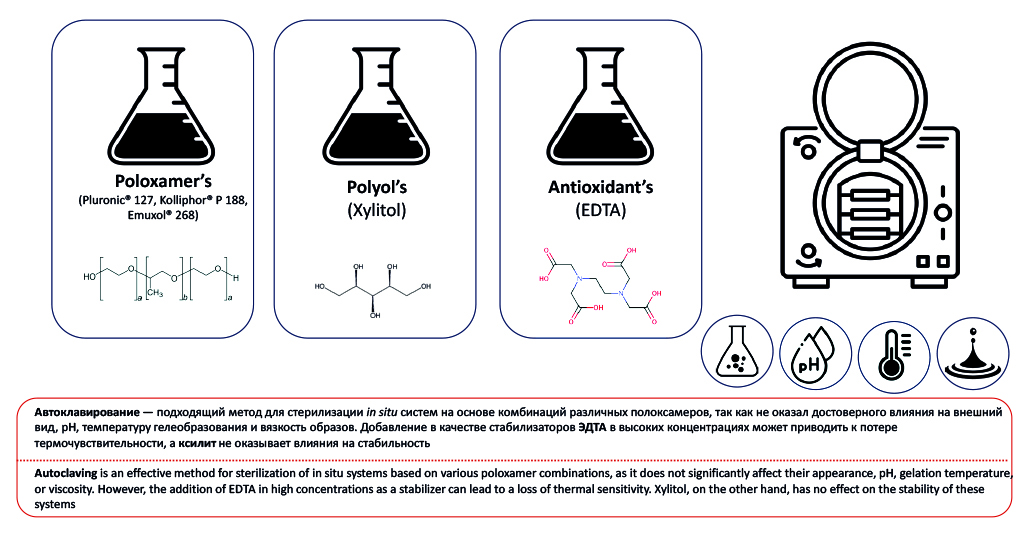
Введение. Парентеральные и офтальмологические in-situ-системы должны быть стерильными. Выбор метода стерилизации – ключевой этап в разработке стерильных стимулочувствительных систем, так как неподходящий метод может привести к разрушению гелеобразующего полимера и активной субстанции и потере активности. К сожалению, стабильность термочувствительных систем на основе полоксамеров при стерилизации, а также способы их стабилизации с помощью протективных агентов изучены недостаточно.
Цель. Целью исследования было изучение стабильности систем на основе полоксамеров при автоклавировании, а также поиск и разработка методов их защиты от негативных последствий стерилизации.
Материалы и методы. В эксперименте использовали полоксамеры Kolliphor® P 407, Kolliphor® P 188, Kolliphor® P 338, Kollisolv® P 124 компании BASF (США), эмуксол-268, проксанол-168, предоставленные компанией АО «НИОПИК» (Россия). В качестве протективных агентов были выбраны динатриевая соль этилендиаминтетрауксусной кислоты (ЭДТА) (ООО ПКФ «ХимАвангард», Россия) и ксилит (ООО «Компания «Сладкий мир», Россия). Образцы автоклавировали при температуре 121 °C в течение 20 мин. Оценка стабильности проводилась по показателям: внешний вид, рН, кинематическая вязкость и температура фазового перехода.
Результаты и обсуждение. Автоклавирование различных комбинаций полоксамеров оказало незначительное влияние на параметры стабильности составов. Добавление ЭДТА в высоких концентрациях приводило к увеличению вязкости, а также снижению рН и гелеобразующей способности составов. После стерилизации во всех образцах с ЭДТА наблюдалось образование осадка геля, однако в течение 5 дней исходный внешний вид составов восстанавливался. Остальные параметры оставались стабильными после автоклавирования. Добавление ксилита оказывало незначительное влияние на исходные показатели полоксамеров, и после стерилизации составы сохраняли стабильность.
Заключение. Результаты проведенных экспериментов показали, что автоклавирование – подходящий метод для стерилизации систем на основе различных комбинаций полоксамеров. Следует избегать добавления ЭДТА, особенно в высоких концентрациях, из-за негативного влияния на ключевые параметры in-situ-систем и риска образования осадка при автоклавировании. Ксилит не нарушает стабильность полоксамеров при стерилизации. В то же время необходимы дальнейшие исследования для оценки потенциала ЭДТА и ксилита в качестве протективных агентов для стабилизации других стимулочувствительных систем.
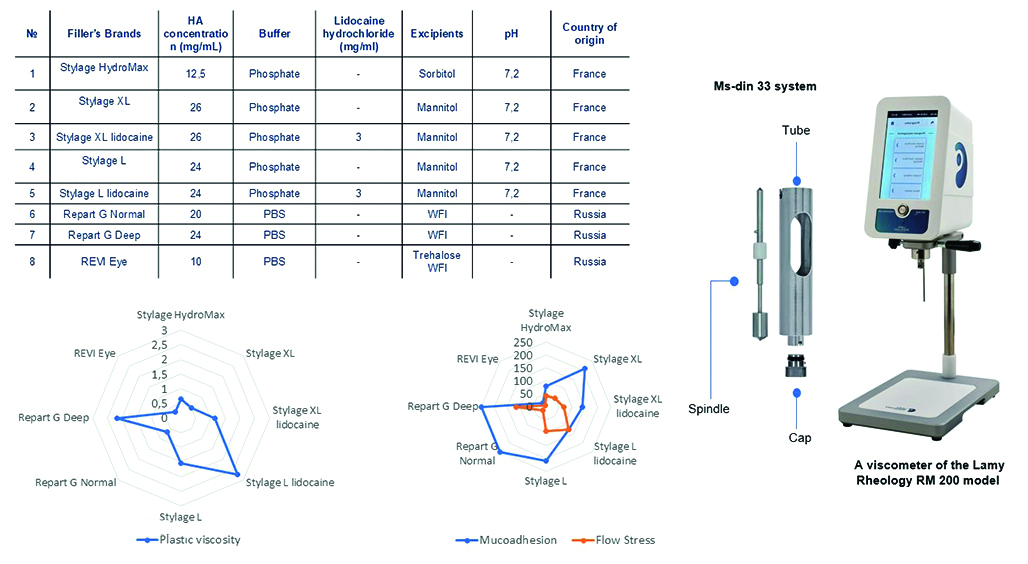
Введение. Гиалуроновая кислота (ГК) – это полисахарид, относящийся к гликозаминогликанам. Благодаря высокой биосовместимости и способности удерживать влагу ГК широко применяется в косметологии и медицине. Популярность гелей на основе ГК растет, особенно в инъекционной косметологии для коррекции возрастных изменений, составляя значительную часть эстетических процедур. В результате ухода иностранных производителей с рынка России появилась необходимость разработки отечественных аналогов. Исследование реологических свойств гелей помогает оптимизировать составы для медицинского применения, обеспечивая эффективность и безопасность новых продуктов.
Цель. Изучить, как концентрация ГК и вспомогательные компоненты влияют на реологические свойства гелей. Также исследование направлено на выявление связи этих свойств с клинической эффективностью.
Материалы и методы. Объекты исследования: филлеры линеек Stylage® – Stylage® HydroMax, Stylage® L, Stylage® L lidocaine, Stylage® XL, Stylage® XL lidocaine, Repаrt® Ġ – Repart G Normal, Repart G Deep. Образцы исследовали методом ротационной вискозиметрии.
Результаты и обсуждение. Была установлена взаимосвязь между клиническими характеристиками жидких имплантов и их реологическими свойствами. В ходе проведенных исследований определены следующие оптимальные диапазоны: мукоадгезия – от 100 до 200 Па, пластическая вязкость – от 0,2 до 1,0 Па ∙ с при скорости сдвига 0–100 с–1 и при температуре 25 ± 1 °С, предел текучести – от 50 до 150 Па при скорости сдвига 0–100 с–1 и при температуре 25 ± 1 °С, тиксотропность – полная.
Заключение. В результате исследование показало, что вязкость важна для инъекционного введения и распределения препарата в тканях. Высокая мукоадгезия в гинекологии и урологии определяет фиксацию имплантата в тканях. Однако чрезмерная мукоадгезивность может вызывать образование подкожных агломератов, что ограничивает использование таких средств на лице. Связь предела текучести с качественными характеристиками имплантов не была установлена.
Введение. Тест сравнительной кинетики растворения (ТСКР) при разработке лекарственных средств является одним из ключевых и моделирует биодоступность действующего вещества в условиях его применения, позволяет оценивать биоэквивалентность лекарственных препаратов. В материалах статьи изложен подход, при котором в процессе разработки за основу для получения лекарственного препарата, отличающегося по лекарственной форме и составу от препарата сравнения, берутся результаты ТСКР.
Цель. Подобрать состав вагинальных таблеток, который будет эквивалентен согласно тесту сравнительной кинетики растворения суппозиториям на липофильной основе.
Материалы и методы. Объектами исследования являются вагинальные таблетки и суппозитории на липофильной основе, содержащие в своем составе натамицин. Исследование проводилось с использованием тестеров растворения (аппарат «Вращающаяся корзинка») и УФ-спектрофотометра для контроля количества высвободившегося натамицина.
Результаты и обсуждение. Авторами был предложен подход, при котором при разработке новой лекарственной формы натамицина (вагинальные таблетки) за основу берутся профили высвобождения зарегистрированных в ЕАЭС вагинальных суппозиториев на липофильной основе. Для проведения ТСКР авторами был получен набор составов вагинальных таблеток. Подбор состава велся таким образом, чтобы получить набор результатов с различными профилями высвобождения и подобрать наиболее оптимальный как с точки зрения эквивалентности профиля по отношению к препарату сравнения, так и технологичности (легкости внедрения в производство и экономичности наработки лекарственного препарата) получаемого состава. Разработанные составы были подобраны таким образом, чтобы обеспечить набор получаемых профилей высвобождения, которые можно было бы в дальнейшем сравнить с референтом. Объекты исследования были проанализированы с помощью тестера растворение (аппарат «Вращающаяся корзинка»), контроль высвободившегося натамицина осуществляли методом УФ-спектрофотометрии.
Заключение. Проведен тест сравнительной кинетики растворения для вагинальных таблеток, содержащих натамицин, и выбран наиболее перспективный состав, отвечающий предъявленным требованиям эквивалентности профилей высвобождения.

Введение. Одной из широких групп лекарств, применяемых в рамках комплексной терапии на разных стадиях поражения печени, являются гепатопротекторы. В клинической практике они применяются в виде отдельных препаратов или в комбинациях. Комбинированный препарат определяется как препарат, содержащий два или больше активных биологических вещества, которые действуют комплексно в организме человека при использовании для профилактики и лечения заболеваний или для восстановления и поддержания состояния здоровья. Комбинированные препараты применяются для повышения эффективности лечения, уменьшения побочных эффектов, упрощения приема лекарственных препаратов или помощи при одновременном лечении нескольких симптомов. Наиболее часто в комбинациях изучаются и применяются следующие хорошо известные гепатотропные средства: эссенциальные фосфолипиды, глицирризиновая кислота, урсодезоксихолевая кислота (УДХК), силимарин. Следует отметить, что хотя фармакологическое действие УДХК и препаратов расторопши изучено хорошо, однако технологических исследований по созданию комбинированных лекарственных средств на их основе не проводилось.
Цель. Разработка технологии получения комбинированного гепатопротекторного средства в виде твердых кишечнорастворимых желатиновых капсул с гранулами, содержащими в качестве активных субстанций УДХК и сухой экстракт расторопши пятнистой в виде твердой дисперсной системы.
Материал и методы. В качестве субстанций в состав разрабатываемого комбинированного препарата гепатопротекторного действия входит УДХК и твердая дисперсная система сухого экстракта расторопши (ТДС СЭР). Твердые желатиновые кишечнорастворимые капсулы наполняли гранулами, содержащими ТДС СЭР и УДХК, с помощью настольной ручной капсулонаполняющей машинки ProFiller 3600, размер капсул – 2. Было наработано 3 серии твердых желатиновых капсул с гранулами ТДС СЭР и УДХК. Проведена стандартизация капсул в соответствии с Государственной фармакопеей Российской Федерации (ГФ РФ) XV издания, ОФС.1.4.1.0005 «Капсулы», по показателям: описанию, подлинности, количественному содержанию суммы флаволигнанов в пересчете на силибин, количественному содержанию УДХК, однородности массы, распадаемости и растворению.
Результаты и обсуждение. При разработке состава гранул с ТДС СЭР и УДХК методом влажной грануляции в качестве вспомогательных веществ использовали микрокристаллическую целлюлозу (МКЦ) или крахмал как дезинтегратор, магния стеарат как лубрикант, в качестве связующего вещества – 5%-й водный раствор картофельного крахмала. Сравнивали полученные составы по показателям качества и по технологическим свойствам. Было установлено, что гранулы, в состав которых входит МКЦ, имеют лучшие технологические свойства: потерю в массе при высушивании, степень гигроскопичности, насыпную плотность, индекс Карра, коэффициент Хауснера, сыпучесть. Для капсул с гранулами ТДС СЭР и УДХК определены показатели качества, они стандартизованы в соответствии с ОФС «Капсулы» по показателям: описанию, подлинности, количественному содержанию суммы флавоноидов в пересчете на силибин, количественному содержанию УДХК, однородности массы дозированных лекарственных форм, распадаемости, растворению. При выполнении теста «Растворение» определяли высвобождение силибина и УДХК к 30, 45 и 60 мин. Высвобождение силибина контролировали спектрофотометрически при длине волны λ = 289 нм, высвобождение УДХК – методом ВЭЖХ в соответствии с фармакопеей США USP 44 – NF 39. Установлено, что за 45 мин высвободилось 89,0 ± 0,5 % силибина и 97,0 ± 0,3 % УДХК.
Заключение. Разработан состав гранул с ТДС СЭР и УДХК, где в качестве дезинтегранта использована МКЦ, лубриканта – магния стеарат, связующего вещества – 5%-й водный раствор картофельного крахмала. Определены технологические свойства гранул и показатели качества. Разработана технология получения кишечнорастворимых капсул, наполненных гранулами, содержащими ТДС СЭР и УДХК. Определены показатели качества, и капсулы стандартизованы по ОФС «Капсулы» по показателям: описанию, подлинности, количественному содержанию суммы флавоноидов в пересчете на силибин (методом УФ спектроскопии), количественному содержанию УДХК (методом ВЭЖХ), однородности массы дозированных лекарственных форм, распадаемости, растворению. Изучены профили высвобождения активной фармацевтической субстанции из кишечнорастворимых капсул и установлено, что к 45 мин высвободилось 89,0 ± 0,5 % суммы флаволигнанов в пересчете на силибин и 97,0 ± 0,3 % УДХК, что соответствует требованиям ГФ РФ XV.
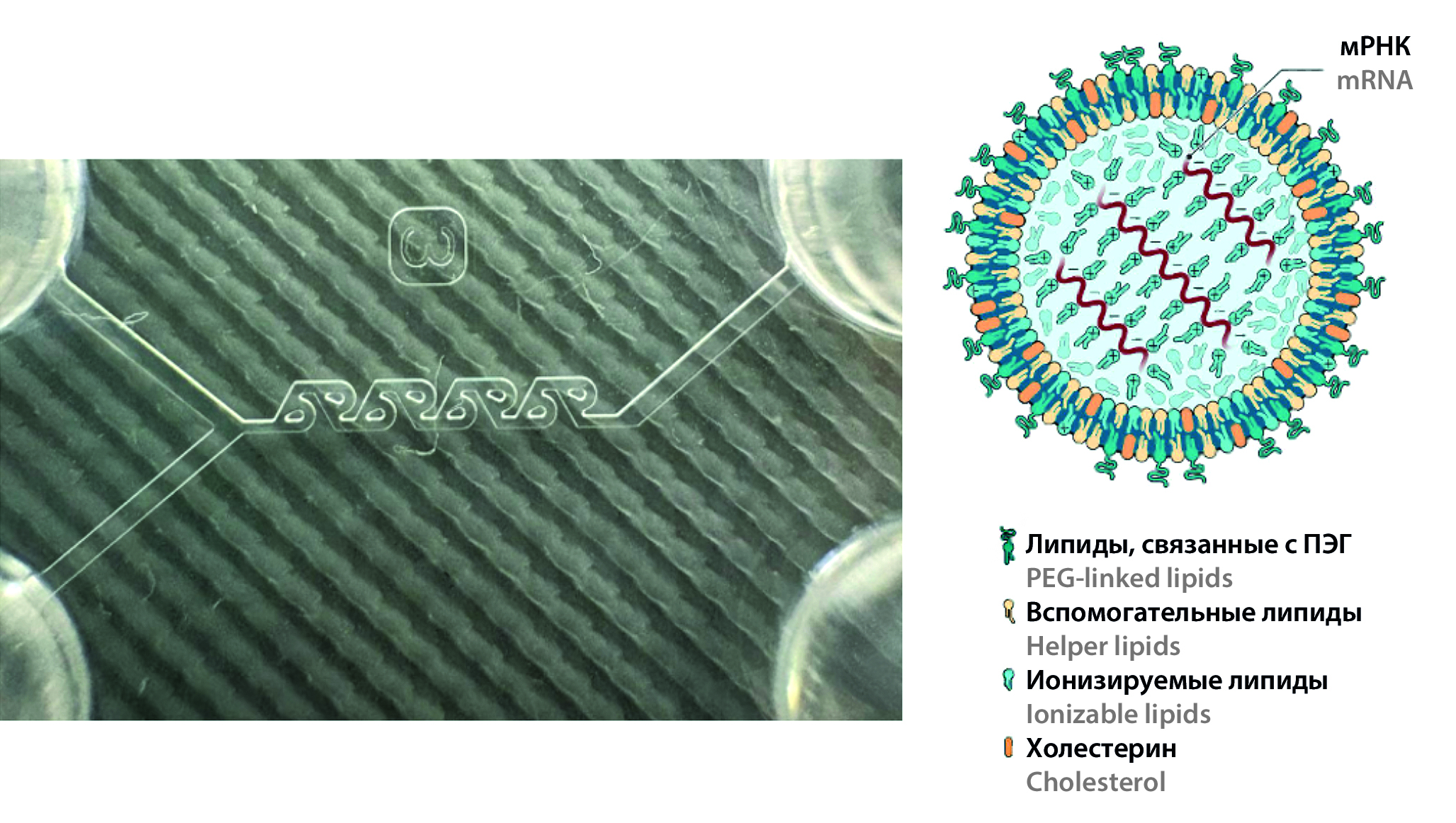
Введение. Генная терапия активно развивается благодаря использованию мРНК-агентов для лечения и профилактики различных заболеваний. Для проявления терапевтического эффекта необходимо доставить мРНК в клетки-мишени и вызвать синтез целевых белков. Основные задачи включают разработку безопасных и эффективных систем доставки. Критическими показателями качества для липидных наночастиц (ЛНЧ) являются средний размер частиц, индекс полидисперсности и значение ζ-потенциала.
Цель. Изучение и оптимизация условий сборки липидных наночастиц для управления их основными характеристиками.
Материалы и методы. Ионизируемый липид гептадекан-9ил(Z)-N-((4-диметиламино)бутил)тио)карбонил)-N-(2-(нон-2-ен-1-илокси)-2-оксоэтил)глицинат (ИЛ) и хелперные липиды – дипальмитоилглицерофосфат (DPPC), холестерол и a-(3’-[1,2-ди(миристилокси)пропанокси]карбониламино}пропил)-w-метоксиполиоксиэтилен (DMG-PEG2000). Растворители: абсолютный спирт, вода очищенная. Буферные растворы: ацетатный буферный раствор (рН 4,5), фосфатный буферный раствор (рН 7,4). Оборудование: микрофлюидная установка Dolomite (Dolomite Microfluidics, Великобритания), Y-образный полимерный микрофлюидный чип с пассивным микромиксером типа «катушка Тесла», анализатор наноразмерных частиц Nanosizer Zeta Pro (ООО «Микротрак», Россия).
Результаты и обсуждение. В рамках исследования было изучено воздействие критических параметров процесса, таких как общая скорость потока (ОСП) и соотношение скоростей потоков (ССП) на свойства ЛНЧ. Были проанализированы такие характеристики, как гидродинамический диаметр (Z-average), средний диаметр частиц (D50), индекс полидисперсности (ИПД, PDI) и ζ-потенциал. Исследование подтверждает, что при увеличении ССП средний гидродинамический и медианный размер частиц уменьшается. Увеличение ОСП также уменьшает гидродинамический и медианный размер частиц за счет гидродинамической фокусировки, но при ОСП = 3200 мкл/мин размер частиц увеличивается из-за снижения стабильности наноэмульсии и агрегации мелких частиц. В исследовании не выявлена прямая зависимость между FRR и ИПД. Наноэмульсии с ССП 1 : 3 и 1 : 4 показали наибольшую однородность. При увеличении ОСП полидисперсность наночастиц снижается. В исследовании не обнаружена зависимость ζ-потенциала липидных наночастиц от оцениваемых параметров процесса. Однако с увеличением ОСП наблюдается тенденция к повышению ζ-потенциала.
Заключение. В исследовании были оптимизированы условия сборки липидных наночастиц (ЛНЧ) с помощью микрофлюидного метода. Предложены математические модели для управления характеристиками наночастиц. Оптимальные условия получения ЛНЧ: ССП = 1 : 3; 1 : 4; ОСП от 2000 до 3000 мкл/мин. При использовании ССП = 1 : 5 и/или ОСП > 3000 мкл/мин показатели могут выйти за пределы оптимального диапазона, что требует дополнительной оценки рисков.
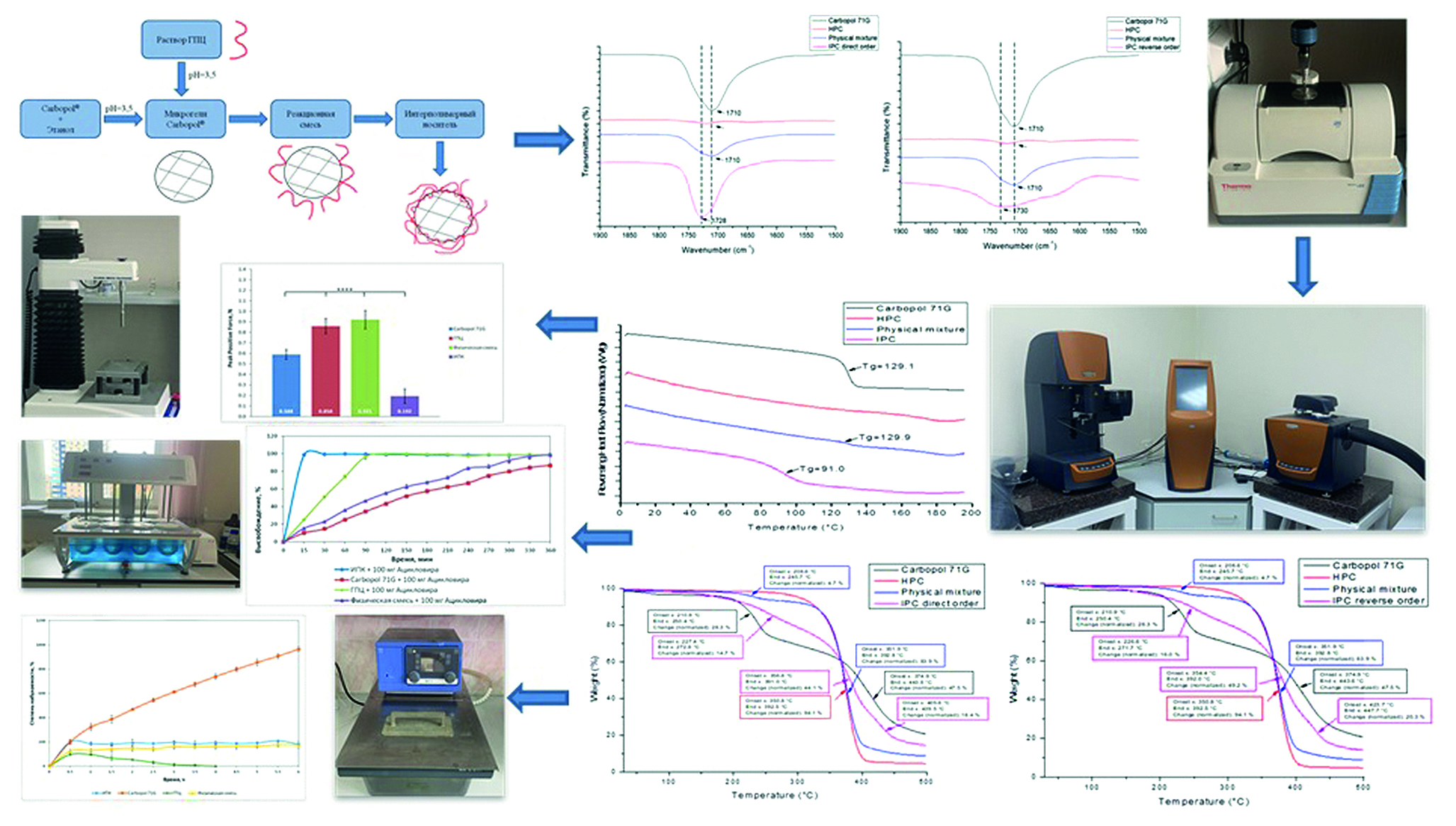
Введение. В результате исследования был изучен процесс образования интерполимерного комплекса (ИПК) между парами полимеров: гидроксипропилцеллюлозой (ГПЦ) и разными марками Carbopol® (71G, 971, 974) – при двух порядках смешения в среде этанола 95 % при pH = 3,5 (подкисленной 0,1 M HCl) методами турбидиметрии, ИК-спектроскопии, термогравиметрического анализа (ТГА). Проведенные эксперименты подтвердили образование поликомплекса между данными парами полимеров. По результатам исследований было выбрано оптимальное соотношение пары ГПЦ и Carbopol® 71G в стехиометричном эквимольном соотношении независимо от порядка их смешивания. С использованием метода модулированной дифференциально-сканирующей калориметрии (мДСК), была подтверждена совместимость изучаемых полимеров в составе образующегося поликомплекса. Согласно проведенному элементному анализу полученный ИПК имел стехиометрический состав ГПЦ / Carbopol® 71G 1 : 2 (по молям). Исследование набухаемости матриц, полученных на основе синтезированного ИПК, а также компактов из индивидуальных полимеров и их физической смеси состава, аналогичного поликомплексу, было проведено в среде, имитирующей желудочных сок, в сравнении с исходными компонентами. Изучение высвобождения ацикловира из полученных носителей также проводилось в среде 0,1 М HCl, результаты показали перспективность разработанной системы для создания носителя с направленным высвобождением лекарственных средств (ЛС) в модельную, имитирующую голодный желудок среду. Исследуемые образцы ИПК показали низкие мукоадгезивные свойства по сравнению с индивидуальными полимерами и их физической смесью. Разработка новых носителей для доставки ЛС является одним из ключевых направлений фармацевтической технологии. В связи с этим особое внимание уделяется веществам полимерной природы, носители на основе которых обеспечивают снижение побочных эффектов, повышение биодоступности и пролонгирования действия ЛС. Системы для гастроретентивной доставки представляют интерес при разработке новых лекарственных форм (ЛФ), позволяющих регулировать скорость высвобождения активного фармацевтического ингредиента (АФИ) в желудке.
Цель. Разработка поликомплексного носителя на основе гидроксипропилцеллюлозы с участием Carbopol® для гастроретентивной доставки ацикловира.
Материалы и методы. Подбор условий образования ИПК проводился с использованием методов турбидиметрии, ИК-спектроскопии, ТГА. Полученный оптимальный состав поликомплексного носителя был охарактеризован с использованием методов мДСК и ИК-спектроскопии. Изучение набухаемости матриц на основе синтезированного ИПК проводилось в среде, имитирующей желудочных сок. Изучение высвобождения ацикловира из полимерных матриц в среду растворения проводили по методу 1 «Вращающаяся корзинка» согласно Государственной фармакопее РФ XV издания. Мукоадгезия исследовалась на анализаторе текстуры TA.XTplus (Stable Micro Systems, Великобритания) на компактах из муцина.
Результаты и обсуждение. Формирование ИПК происходит посредством образования водородных связей между —OH-группами макромолекулярных звеньев линейной ГПЦ и —COOH-группами редкосшитой полиакриловой кислоты (рПАК) в составе используемых марок карбополов. Смещение характеристической полосы влево до 1730 см–1 на ИК-спектрах поликомплексов подтверждает образование ИПК. ИПК характеризуются единственной температурой стеклования (Тс = 91,0 ± 2,1 °C). Элементный анализ выявил двухкратный мольный избыток редкосшитого полимера (Carbopol® 71G) над линейным (ГПЦ). На протяжении всего эксперимента по изучению кинетики набухаемости компактированные матрицы сохраняют свою форму, увеличиваясь в размерах. С использованием термического анализа образцов поликомплексных матриц в процессе оценки их набухаемости был проведен мониторинг возможных структурных преобразований, подтвердивший устойчивость поликомплекса в кислой среде. По результатам исследования кинетики высвобождения модельного АФИ максимальная концентрация ацикловира, перешедшего в среду, наблюдается на 30-й мин эксперимента и составляет 98,5 %. Тогда как для ГПЦ-матриц максимальная концентрация достигается по истечении 2 ч, а для матриц на основе Carbopol® 71G и их физической смеси (ФС) – только в заключительной части эксперимента. Синтезированный ИПК характеризовался низкой способностью к мукоадгезии к компактам из муцина по сравнению с индивидуальными полимерами и ФС.
Заключение. В результате исследования были подобраны оптимальные условия образования ИПК между парами полимеров – ГПЦ и изучаемыми марками Carbopol® (71G, 971 и 974). Методами турбидиметрии, ИК-спектроскопии и ТГА доказано образование поликомплексов на основе ГПЦ и различных марок Carbopol®. ИПК ГПЦ / Carbopol® 71G стехиометрического состава, подтвержденный элементным анализом, был охарактеризован с использованием термических и спектральных методов. Изучение высвобождения ацикловира из полученных матриц показало перспективность применения разработанной системы для пероральной гастроретентивной его доставки.

Введение. Благодаря широкому спектру биологической активности транс-ресвератрол является перспективным кандидатом для создания лекарственных препаратов на его основе. Однако низкая растворимость в воде и химическая нестабильность субстанции при пероральном приеме ограничивает его применение в клинической практике. В связи с этим перспективно рассмотреть альтернативные системы доставки, лимитирующие эффект первого прохождения через печень.
Цель. Разработка и фармакотехнологическая оценка трансдермальных пластырей с ресвератролом.
Материалы и методы. Объект исследования – субстанция транс-ресвератрола (DSM, Швейцария). В качестве полимеров-носителей, обеспечивающих адгезию пластырей, рассматривали поливинилпирролидон (ПВП) различной молекулярной массы (К-17, К-30, К-90, USP, Dalian Sinobio Chemistry Co., Ltd., Китай) и сополимер метакриловой кислоты и этилакрилата (BASF, Германия). Роль пластификатора выполнял полиэтиленгликоль-400 (ПЭГ-400) (ООО ГК «РусХим», Россия). Натрия метабисульфит (Yantai Sodium Metabisulphite Co., Ltd., Китай) использовали как антиокислитель, а спирт этиловый 95%-й (ФС.2.1.0036, Р N003960/01, ООО «РОСБИО», Россия) – как растворитель компонентов матрицы. Полиэтилентерефталатная пленка толщиной 20 мкм была необходима для создания внешнего покровного слоя (подложки), антиадгезивная силиконизированная бумага – для защиты матриц. Пластыри готовили методом полива. Высушивание проводили в климатической камере HPP110 (Memmert, Германия). В рамках контроля качества готовых ТТС, согласно требованиям методики «FTM 8» руководства международной ассоциации FINAT, оценивали сопротивление сдвигу, а также проводили оценку липкости в соответствии с монографией «Methods of Adhesion Testing» Японской фармакопеи 18-го издания. Роль препарата сравнения выполнял трансдермальный пластырь Никоретте® (LTS Lohmann Therapy-Systems AG, Германия). Для изучения биофармацевтических свойств разработанных составов использовали тестер растворения ERWEKA DT 626 (ERWEKA GmbH, Германия) с диском-держателем. Оценку гигроскопичности матриц проводили при помощи сушильного шкафа BINDER FED 53 (BINDER GmbH, Германия). Результаты исследования анализировали стандартными методами статистики в соответствии с требованиями Государственной фармакопеи РФ. Для сравнения показателей адгезии между группами проводили однофакторный дисперсионный анализ (One-way ANOVA, GraphPad Prism 8.0.2, США) при p < 0,0001.
Результаты и обсуждение. При сравнительной оценке сопротивления сдвигу было установлено, что увеличение толщины матрицы приводит к росту числа сдвиговых слоев, что снижает ее когезионную прочность. Введение высокомолекулярных ПВП К-30 и К-90 в рецептуру к ПВП К-17 обеспечивает концентрационно зависимое увеличение внутренней прочности композиции и повышает ее сопротивление сдвиговым деформациям, при этом оказывает негативное влияние на высвобождение действующего вещества из полимерной матрицы. Состав на основе сополимера метакриловой кислоты и этилакрилата иллюстрирует оптимальное сочетание адгезивных и биофармацевтических свойств.
Заключение. Исследование подтверждает, что разработка ТТС представляет собой сложный, многоэтапный процесс, требующий сбалансированного подхода к оптимизации состава. Критически важным аспектом является необходимость комплексной оценки нескольких ключевых показателей качества, поскольку модификация рецептуры, направленная на улучшение одних характеристик, может привести к ухудшению других. В рамках дальнейшей разработки пластырей с ресвератролом перспективно использовать матрицу на основе сополимера метакриловой кислоты и этилакрилата, а также рассмотреть возможность совершенствования состава на основе ПВП К-17 с целью улучшения его адгезионных характеристик.
МЕТОДЫ АНАЛИЗА ЛЕКАРСТВЕННЫХ СРЕДСТВ
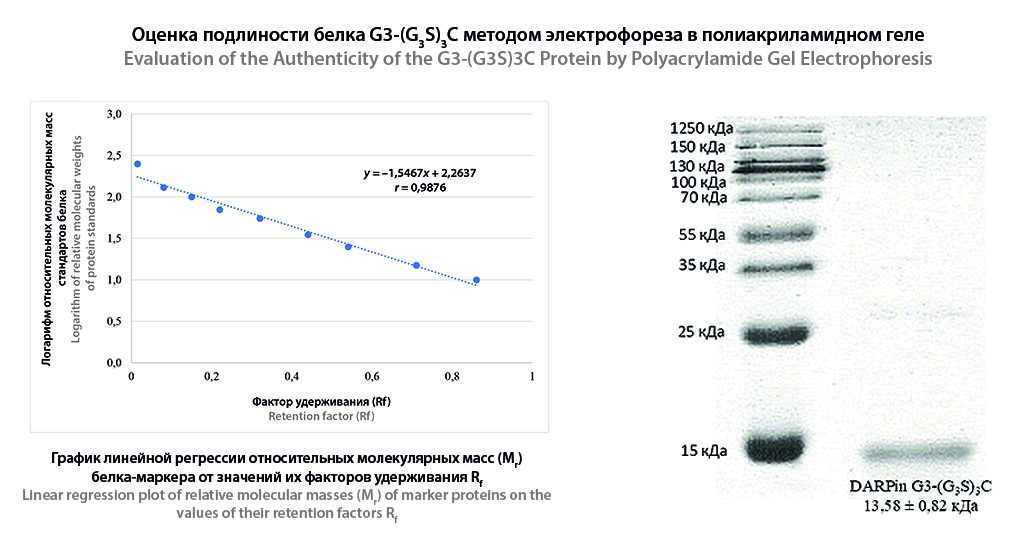
Введение. Сверхэкспрессия рецептора HER2 ассоциирована с агрессивным течением онкологических заболеваний и неблагоприятным прогнозом. Традиционная иммуногистохимическая диагностика имеет ряд ограничений, включая инвазивность, невозможность оценки гетерогенности опухоли и тотального распространения процесса. Перспективной альтернативой является радионуклидная визуализация с использованием каркасных таргетных белков DARPin G3. По результатам доклинических исследований препарат 99mTс[Tc]-G3-(G3S)3C был предложен для проведения I фазы клинических исследований. Экспериментальный препарат представляет собой стерильный лиофилизат химического предшественника в одном флаконе и должен соответствовать требованиям ОФС 1.11.0005 «Химические предшественники для радиофармацевтических лекарственных препаратов» и ОФС 1.7.1.0007.15 «Лекарственные средства, получаемые методами рекомбинантных ДНК» Государственной фармакопеи РФ XV издания.
Цель. Целью настоящего исследования была разработка подходов и методик контроля качества белка DARPin G3-(G3S)3C, входящего в состав разработанной композиции лиофилизата химических предшественников для получения 99mTс-содержащего препарата для радионуклидной визуализации гиперэкспрессии HER2 в злокачественных опухолях
Материалы и методы. Объектом исследования явился лиофилизат, содержащий DARPin G3-(G3S)3C со вспомогательными веществами. Для регистрации масс-спектров белка использовали ВЭЖХ с тандемным масс-спектрометром, снабженным источником ионизации электрораспылением. Вертикальный электрофорез с натрия додецилсульфатом (SDS) в полиакриламидном геле с концентрацией акриламида 15 % проводили при напряжении 110 В в течение 2 ч. В качестве красящего раствора использовали раствор кумасси В-250. Тандемную масс-спектрометрию проводили с помощью системы ВЭЖХ с масс-спектрометром посредством наноэлектроспрейного источника. При корреляции аминокислотной последовательности карбамидометилирование Cys, деамидирование Asn/Gln и окисление Met учитывались как вариабельные модификации. Для определения количества белка в лиофилизате был использован спектрофотометрический метод при длине волны 280 нм. Валидационная оценка разработанной методики проводилась в соответствии с требованиями ОФС.1.1.0012 «Валидация аналитических методик» Государственной фармакопеи РФ XV издания.
Результаты и обсуждение. Подлинность и чистота DARPin G3-(G3S)3C была подтверждена с использованием ВЭЖХ-МС. Молекулярная масса мономера согласуется с расчетной и составила 14075,9 Да с точностью до 0,5 Да, чистота белка была близка к 100 %. Для определения подлинности и чистоты DARPin G3-(G3S)3C методом электрофореза в полиакриаламидном геле предложено использовать 2 мкг и 20 мкг белка соответственно. Рассчитанная молекулярная масса белка составила 13,58 ± 0,82 кДа. В лиофилизате содержание примеси гомодимера, определенной электрофорезом в полиакриламидном геле, не превышало 3 %, другие белковые примеси наблюдались в количестве не более 1 %. Чистота белка, установленная методом ВЭЖХ-МС/МС для анализа первичной структуры, составила 98 %. Перекрытие аминокислотной последовательности целевого белка с идентифицированными пептидными фрагментами составило 100 %. В результате анализа гидролизата белка было идентифицировано более 700 его пептидных фрагментов (значение –10LоgP – более 20). Разработанная методика УФ-спектрофотомерии соответствует валидационным требованиям и позволяет определять количественного содержание белка DARPin G3-(G3S)3C в лиофилизате ±10 % от номинального содержания.
Заключение. Для определения показателя «подлинность» нового химического предшественника белка DARPin G3-(G3S)3C в составе лиофилизата для получения 99mTс-содержащего препарата для радионуклидной визуализации гиперэкспрессии HER2 в злокачественных опухолях были предложены методики масс-спектрометрии, пептидного картирования, электрофорез в полиакриламидном геле. Для показателя «родственные примеси» – методика электрофореза в полиакриламидном геле. Для определения показателя «количественное определение» – методика УФ-спектрофотометрии.
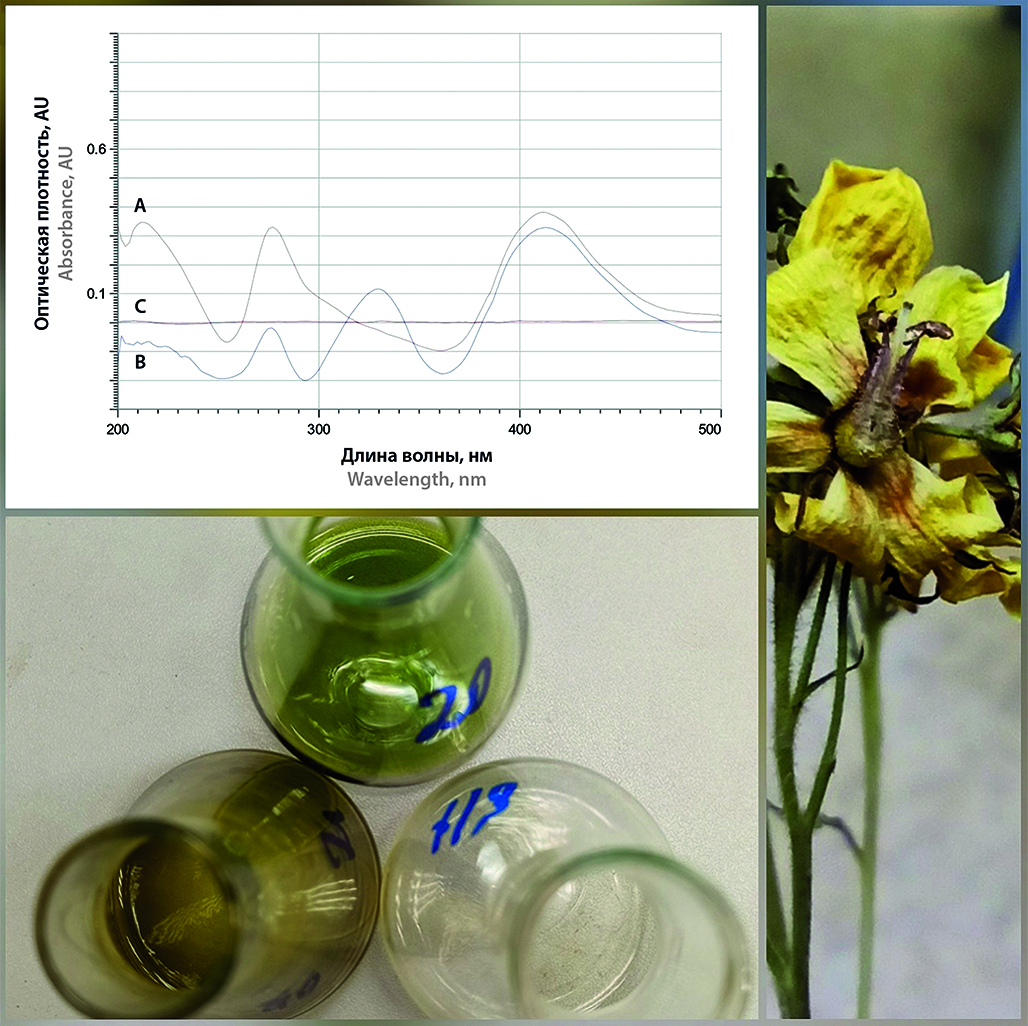
Введение. Исследование качественного и количественного состава растений и выявление перспективных лекарственных видов – одна из важнейших задач современной фармакогнозии. Вербейник обыкновенный (Lysimachia vulgaris L.) сем. первоцветных (Primulaceae Batsch ex Borkh.) – многолетнее травянистое растение, для надземной части которого основной группой действующих веществ являются фенольные соединения. L. vulgaris распространен в России практически повсеместно, известно о его применении в традиционной медицине, а также о видах биологической активности, рассмотренных в экспериментах in vitro и in vivo, но поиск научной литературы в отечественном сегменте не выявил достаточной информации о количественном содержании биологически активных веществ (далее – БАВ).
Цель. Разработка и валидация методики оценки количественного содержания суммы флавоноидов в траве вербейника обыкновенного (L. vulgaris) в пересчете на рутин методом спектрофотометрии.
Материалы и методы. В качестве объекта использовали спиртовые извлечения из надземной части L. vulgaris, заготовленного в период цветения в Ленинградской области. Количественное определение проводили методом спектрофотометрии. Предложенную методику валидировали в соответствии с ОФС.1.1.0012 «Валидация аналитических методик» Государственной фармакопеи РФ XV издания по показателям «специфичность», «линейность», «аналитическая область», «повторяемость», «прецизионность» и «правильность».
Результаты и обсуждение. Для методики количественного определения флавоноидов в траве L. vulgaris методом спектрофотометрии в качестве стандартного образца был предложен рутин. Было установлено, что оптимальное извлечение флавоноидов из травы L. vulgaris достигается путем использования однократной экстракции спиртом этиловым 70%-м в течение 30 мин при соотношении «сырье : экстрагент» 1 : 100 и измельченности сырья 2–1 мм. В качестве комплексообразователя был предложен раствор алюминия хлорида 1%-й спиртовой, были подобраны условия комплексообразования (аликвота раствора алюминия хлорида 1%-го – 2 мл, время комплексообразования – 30 мин). Методика отвечает требованиям валидационных характеристик. По разработанной методике было установлено, что сумма флавоноидов в траве L. vulgaris, заготовленного в Ленинградской области, составляет 2,97 ± 0,05 % в пересчете на рутин.
Заключение. Разработана и валидирована методика количественного определения суммы флавоноидов в траве вербейника обыкновенного (L. vulgaris) методом спектрофотометрии в пересчете на рутин.
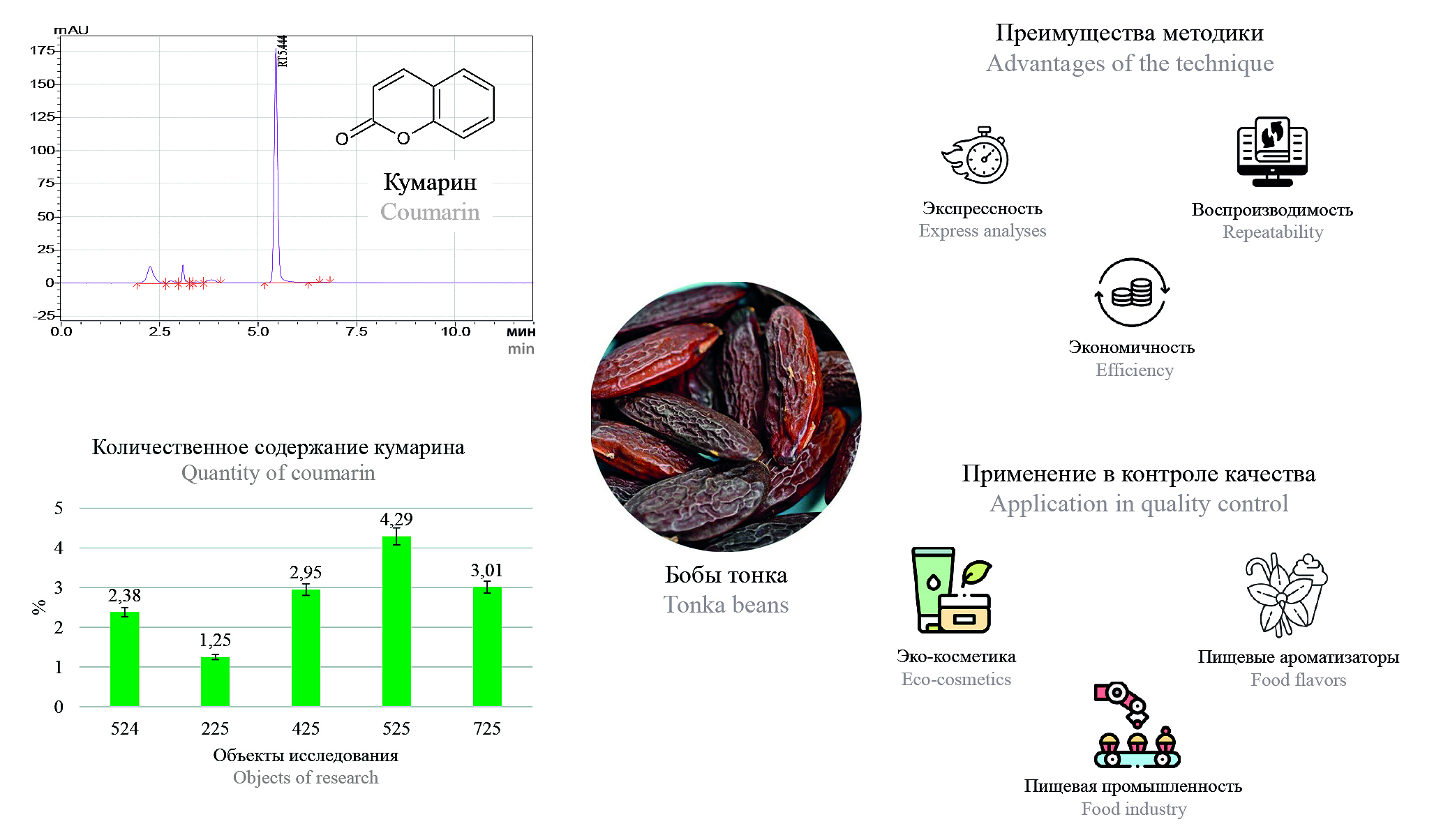
Введение. Бобы тонка (семена Dipteryx odorata, семейство Fabaceae) – ценный источник кумарина, применяемого в пищевой, парфюмерной и косметической промышленности благодаря насыщенному аромату. Несмотря на фармакологическую ценность (антикоагулянтная, антиоксидантная активность), кумарин обладает доказанной гепато- и гематотоксичностью, что ограничивает его использование в ряде стран. Несмотря на контроль синтетического кумарина в ароматизаторах, природный аналог из бобов тонка не имеет четких нормативов, хотя обладает идентичной активностью. Рост спроса на натуральные продукты повысил популярность бобов тонка, однако недостаток научных данных об их безопасности и методах анализа затрудняет контроль качества. Разработка валидированных методик анализа с использованием метода ВЭЖХ – актуальная задача для обеспечения безопасности его применения в промышленности.
Цель. Разработка ВЭЖХ-методики количественного определения кумарина в бобах тонка и ее валидация с дальнейшей апробацией на нескольких партиях исследуемого объекта.
Материалы и методы. В качестве объектов исследования были использованы пять партий бобов тонка различных стран-производителей. Условия хроматографирования: колонка Luna C18(100) Å LC Column, 250 × 4,6 мм, 5 мкм, температура термостата – 40 °С, длина волны детекции – 275 нм, ПФ А – ацетонитрил, ПФ Б – вода. Режим элюирования: изократика (ПФ Б – 50 %) в течение 12 мин.
Результаты и обсуждение. Разработанная методика валидна по показателям пригодности хроматографической системы, специфичности, линейности (диапазон 0,001–0,1 мг/мл) и прецизионности. Ее преимущества – экспрессность и экономичность, что упрощает контроль качества сырья. Количественное содержание кумарина в исследуемых объектах варьируется в диапазоне от 1,25 ± 0,07 % до 4,29 ± 0,12 %, что коррелирует с данными зарубежных исследователей.
Заключение. Разработана и валидирована ВЭЖХ-методика для количественного определения кумарина в бобах тонка. Разработанная методика может быть использована для стандартизации и рутинного анализа этого перспективного сырья в различных отраслях промышленности, обеспечивая необходимый уровень безопасности при его применении.

Введение. Кумарин и его производные – БАВ растительного происхождения, проявляющие некоторые виды фармакологической активности, а также токсикологические эффекты. Побочные действия препаратов – производных кумарина, а также отравления, возникающие в связи с применением крысиного яда, кумаринсодержащих растений и т. д., обосновывают актуальность разработки экспрессной методики анализа производных кумарина.
Цель. Разработать экспрессную методику полуколичественного определения производных кумарина с применением бумажной хроматографии, провести валидационные испытания и апробацию методики на растительных объектах.
Материалы и методы. Для разработки методики использовали субстанции кумарина и гидроксида натрия, фильтровальную бумагу марки ФС и УФ-фонарь (365 нм). Методика включает импрегнацию бумажных полосок растворами гидроксида натрия (5–30 %), пробоподготовку образцов экстракцией водой или этанолом с последующей фильтрацией и визуальную детекцию флуоресценции. Валидацию проводили с использованием стандартных образцов фенольных соединений (производные кумарина, гидроксикоричных, бензойных кислот и флавоноидов).
Результаты и обсуждение. В результате исследований установлены оптимальные условия методики: концентрация импрегнирующего раствора гидроксида натрия – 10 %, использование бумаги марки ФС, время детекции – 20 с. Показано, что методика обладает селективностью к производным кумарина при выполнении ряда испытаний, которые отражены в дереве принятия решений. Подтверждена возможность полуколичественного определения с пределом обнаружения 1 · 10–6 мг/мл, проведена апробация на синтетических и растительных объектах.
Заключение. Разработана экспресс-методика полуколичественного определения производных кумарина, основанная на их щелочном гидролизе на бумажной подложке с детекцией по флуоресценции. Методика отличается простотой, относительно высокой чувствительностью (предел обнаружения – 1 · 10–6 мг/мл) и была успешно апробирована на лекарственных средствах, растительном сырье и других объектах. Предлагаемая методика рекомендована для использования в качестве эффективного инструмента предварительного скрининга в лабораторной практике.
ДОКЛИНИЧЕСКИЕ И КЛИНИЧЕСКИЕ ИССЛЕДОВАНИЯ

Введение. Физиологически обоснованное моделирование фармакокинетики представляет собой метод, позволяющий предсказывать распределение лекарственных веществ в организме на основе анатомических и физиологических параметров. Данное направление получило широкое распространение лишь с развитием вычислительных технологий. Сегодня PBPK активно используется регуляторными агентствами для оптимизации клинических исследований и сокращения числа экспериментов на животных.
Текст. PBPK-модели представляют организм как систему взаимосвязанных компартментов, соответствующих органам и тканям. Описываются три основных типа моделей: полноценная (Full PBPK), обеспечивающая максимальную точность за счет детализации; усеченная (Reduced PBPK), снижающая вычислительную сложность; и гибридная (Hybrid PBPK), сочетающая оба подхода для баланса между точностью и эффективностью. Подробно рассмотрены ключевые параметры для построения моделей: физико-химические свойства веществ (LogP, pKa, растворимость), физиологические параметры (объемы органов, скорость кровотока, активность ферментов и мембранных транспортных белков) и фармакокинетические параметры (объем распределения, клиренс). Особое внимание уделяется модели всасывания и транзита Гордона Амидона (CAT/ACAT) и её интеграции в PBPK-моделирование. Приведены процедуры обеспечения надежности моделей: калибровка (настройка параметров), валидация (оценка предсказательной способности), квалификация (подтверждение пригодности для конкретной цели) и верификация (проверка математической корректности). Описаны статистические метрики для оценки точности. Представлен обзор популярного программного обеспечения для PBPK-моделирования, такого как GastroPlus, Simcyp, PK-Sim, SimBiology и Mrgsolve, с указанием их основных преимуществ и областей применения в фармацевтической индустрии и академических исследованиях.
Заключение. PBPK-моделирование находится на пороге новой эры, где его применение выйдет за рамки традиционной фармакокинетики, став неотъемлемой частью цифровой медицины, биотехнологий и прецизионной терапии. В перспективе такие технологии смогут не только предсказывать поведение лекарств в организме, но и стать основой для виртуальных клинических испытаний, что коренным образом изменит подход к разработке и применению лекарственных средств.
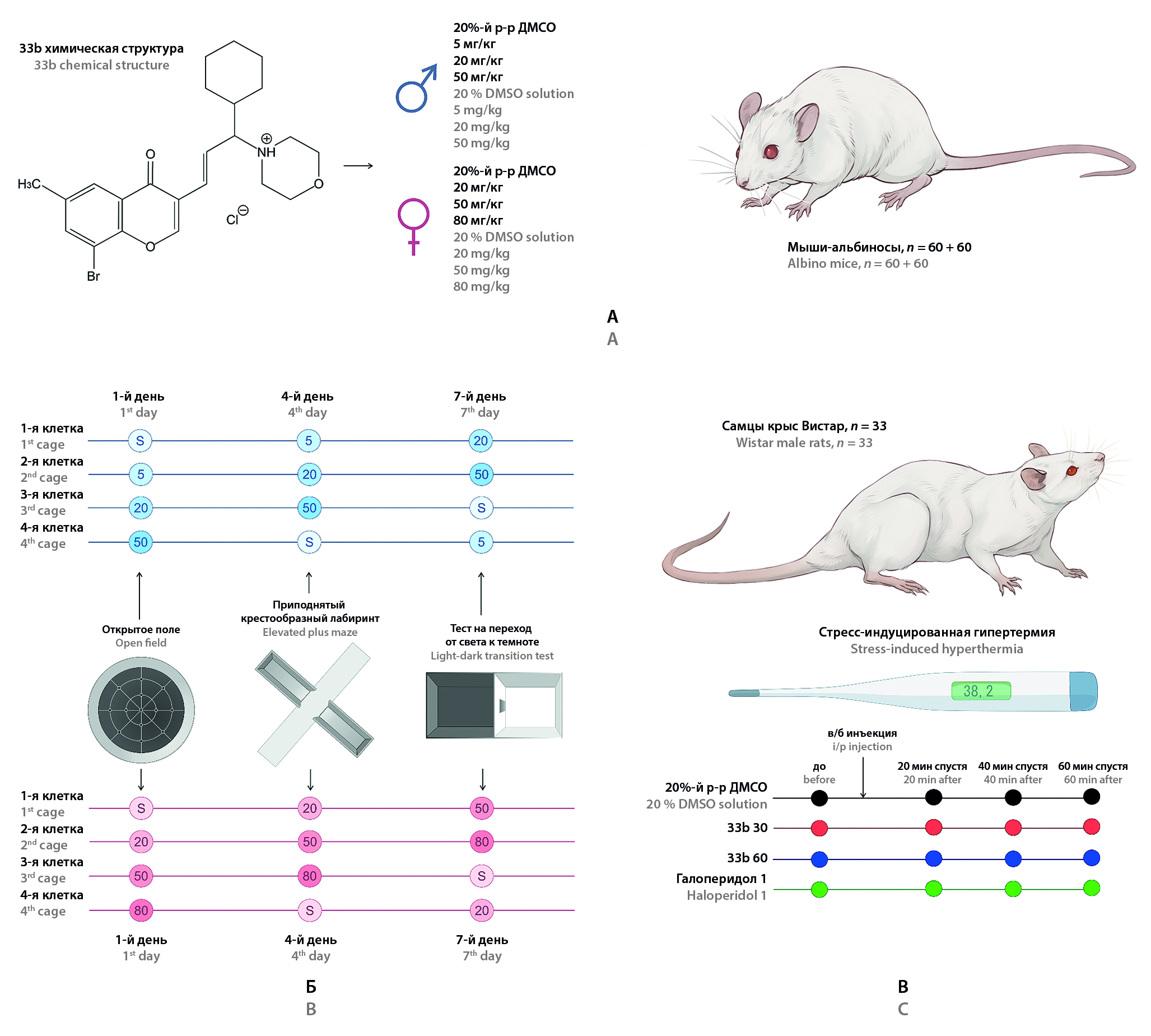
Введение. Хромонсодержащие производные аллилморфолина (ПАМ) – новая группа биологически активных соединений с предполагаемой психотропной активностью. В серии экспериментов на рыбах Danio rerio ПАМ продемонстрировали выраженный дозозависимый седативный эффект, а соединение 33а в малых дозах оказывало анксиолитическое действие в тесте «Новый аквариум». Проведенный методом фармакоэнцефалографии скрининг другой молекулы из ряда ПАМ, 33b, продемонстрировал сходство ее эффектов в отношении ЭЭГ с дофаминоблокатором сульпиридом и антигистаминным средством хлоропирамином. На основании результатов этого прогноза можно предположить у молекулы седативное и антипсихотическое действие.
Цель. Изучение фармакологической активности соединения 33b в тестах на эмоционально-тревожное поведение у мышей, а также в тесте «Стрессиндуцированная гипертермия» у крыс.
Материалы и методы. Оценку фармакологической активности 33b проводили в тестах «Открытое поле» (ОП), «Приподнятый крестообразный лабиринт» (ПКЛ), «Темно-светлая камера» (ТСК) на самцах и самках беспородных белых мышей, а также в тесте «Стрессиндуцированная гипертермия» на самцах крыс линии Wistar.
Результаты и обсуждение. В тестах ОП, ПКЛ и ТСК введение 33b в дозах 50 и 80 мг/кг приводило к стойкому снижению числа стоек и у самок, и у самцов. Снижение локомоторной активности в тесте ОП оставалось на уровне тенденции. Угнетение вертикальной активности у самцов происходило при введении меньших доз 33b, чем у самок. Данных об анксиолитическом действии молекулы в данных тестах получено не было. В тесте «Стрессиндуцируемая гипертермия» у крыс наблюдали выраженное снижение температуры тела при введении 33b в дозе 60 мг/кг.
Заключение. Полученные данные продемонстрировали умеренное седативное действие соединения 33b, при этом чувствительность самцов к действию молекулы была выше. В тесте на крысах для изучаемого ПАМ было показано гипотермическое действие в условиях стресса, что может быть подтверждением его дофаминоблокирующего действия. Это позволяет рассматривать вещество 33b из группы хромонсодержаших аллилморфолинов перспективным соединением для дальнейших исследований в качестве потенциального антипсихотического средства.

Введение. Лекарственные растения, такие как копеечник Семенова (Hedysarum semenowii Regel & Herder) рода Hedysarum L., обладают богатым фитохимическим профилем и могут стать перспективной основой для разработки новых фармацевтических препаратов. Несмотря на известную фармакологическую активность многих видов Hedysarum L., профиль безопасности этих растений изучен недостаточно, что требует дополнительной оценки их токсичности и потенциальных побочных эффектов. В этой связи проведены исследования по определению подострой токсичности, местнораздражающего действия и аллергенности экстракта Hedysarum semenowii для оценки его безопасности и терапевтического потенциала.
Цель. Определение подострой токсичности, местнораздражающего действия и аллергенности экстракта копеечника Семенова (Hedysarum semenowii Regel & Herder).
Материалы и методы. Исследование подострой токсичности экстракта Hedysarum semenowii проводилось на беспородных крысах, где животным вводили экстракт надземной части растения перорально в дозах 500, 2000 и 5000 мг/кг в течение 28 дней, наблюдая за их состоянием. По окончании исследований подострой токсичности исследовали патологические изменения в печени и почках экспериментальных животных. Местнораздражающее и аллергенное действие оценивали на крысах и морских свинках соответственно с использованием визуальных методов и шкалы Дрейза.
Результаты и обсуждение. В ходе 28-дневного исследования подострой токсичности у животных не выявлено значимой токсичности, смертности или изменений в поведении. Масса тела, потребление корма и воды оставались стабильными. По результатам макроскопического и микроскопического исследования в печени и почках отмечены незначительные физиологические изменения, выраженность которых коррелировала с дозой введенного экстракта. Кроме того, экстракт не вызывал раздражения кожи или аллергенных реакций в тестах с участием крыс и морских свинок.
Заключение. В результате исследований экстракт Hedysarum semenowii продемонстрировал низкую токсичность, отсутствие раздражающего и аллергизирующего действий, что подтверждает его безопасность и перспективность для дальнейшего изучения в качестве потенциального терапевтического средства.
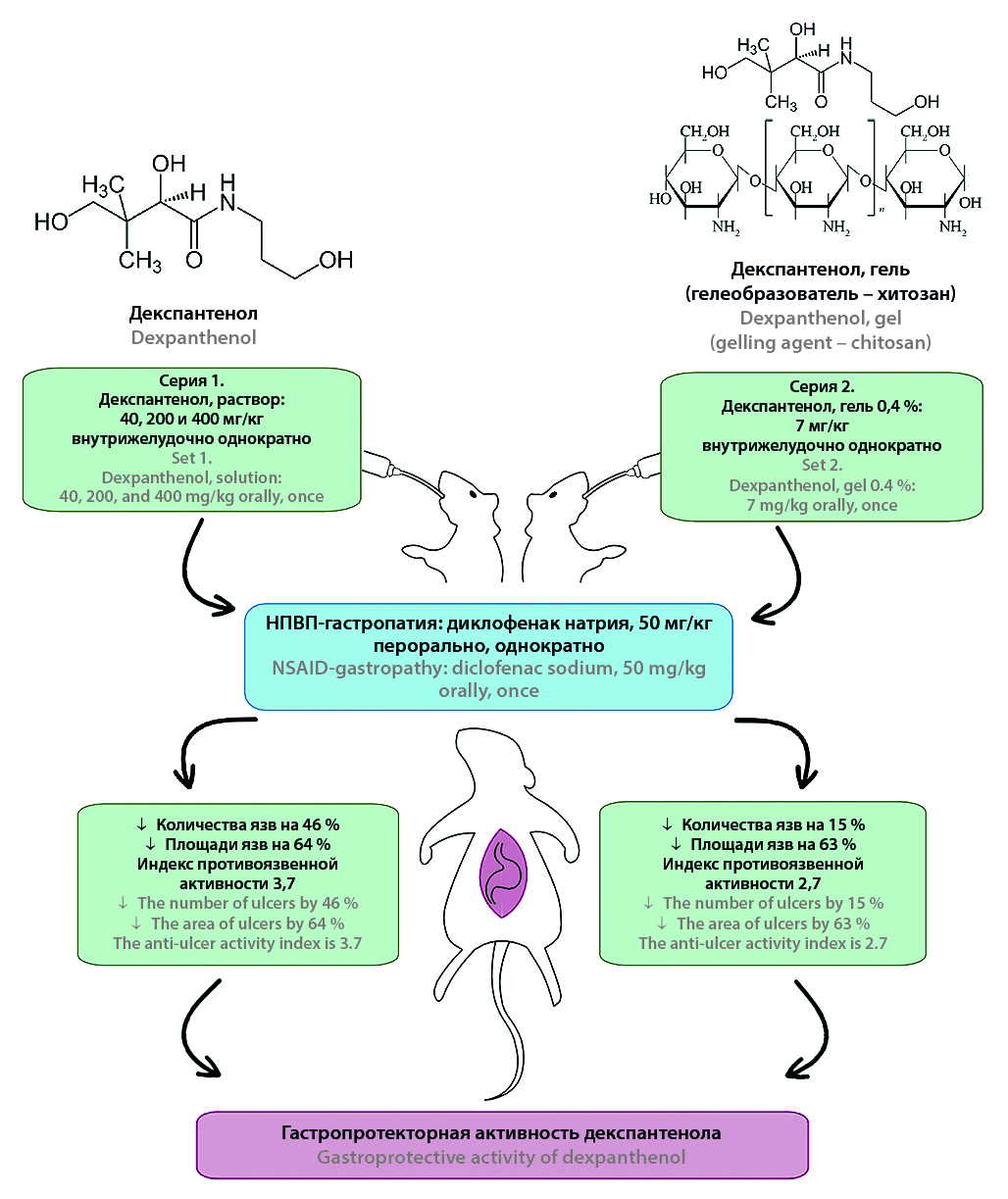
Введение. Актуальным остается поиск новых гастропротекторов, одним из которых может являться декспантенол – предшественник пантотеновой кислоты, нетоксичный при приеме внутрь. В РФ не зарегистрированы препараты декспантенола для системного применения, использование для лечения кислото-зависимых заболеваний (КЗЗ) желудочно-кишечного тракта не известно, перспективно изучение его гастропротекторных свойств при пероральном применении.
Цель. Доклинические исследования гастропротекторной активности декспантенола при приеме внутрь на модели гастропатии, индуцированной нестероидными противовоспалительными препаратами (НПВП-гастропатии).
Материалы и методы. В эксперименте использовано 142 крысы-самца массой 220–280 г (конвенциональные, рандомбредные, альбиносы). НПВП-гастропатию моделировали путем введения диклофенака натрия внутрь однократно в дозе 50 мг/кг, эвтаназию проводили путем передозировки ингаляционного наркоза через 3 ч после введения диклофенака натрия. Декспантенол вводили внутрь однократно за 1 ч до диклофенака натрия: в дозах 400, 200, 40 мг/кг (раствор водный), в дозе 7 мг/кг (гель для приема внутрь 0,4 масс.%, гелеобразователь – хитозан высоковязкий). Препараты сравнения: висмута трикалия дицитрат, 17 мг/кг, диоксометилтетрагидропиримидин (син. метилурацил) в дозе 42 мг/кг – вводили однократно внутрь за 1 ч до диклофенака натрия. При помощи разработанного программного обеспечения [язык Python с графическим пользовательским интерфейсом (GUI)] определяли площадь язв на слизистой оболочке желудка (СОЖ) и их количество. Вычисляли по известным формулам индексы Паулса и противоязвенной активности, проводили гистологические исследования СОЖ, окраска – гематоксилин-эозин.
Результаты и обсуждение. При применении декспантенола выявлено дозозависимое уменьшение площади язвенных дефектов на СОЖ по сравнению с контролем, максимально в 2,7 раза (в дозе 40 мг/кг, 1,0 масс.% водный раствор, однократно перорально), значение индекса противоязвенной активности – 3,7, что выше, чем эффективность метилурацила. Гель для приема внутрь 0,4 масс.%, содержащий декспантенол, в дозе 7 мг/кг обеспечивает уменьшение площади язвенных дефектов на СОЖ на 63 %, величина индекса противоязвенной активности – 2,7, что превышает эффективность висмута трикалия дицитрата, подтверждается морфологическими признаками уменьшения выраженности воспалительного процесса, активизацией регенерации и, вероятно, обусловлено не только активностью декспантенола, но и синергизмом с хитозаном, а также обволакивающим действием лекарственной формы гель. Выявленная гастропротекторная активность декспантенола согласуется с известными механизмами его действия, в том числе такими, как сохранение целостности фосфолипидного бислоя, влияние на синтез муцина и белков плотных контактов (TJ), ядерные факторы транскрипции и апоптоза, факторы роста, цитокины, медиаторы воспаления, деление и дифференцировку клеток.
Заключение. Впервые доказано, что декспантенол обладает гастропротекторной активностью при приеме внутрь, что проявляется значительным достоверным уменьшением количества и площади язвенных дефектов СОЖ на модели НПВП-гастропатии при сравнении с контрольной группой, индекс противоязвенной активности: 3,7 – в дозе 40 мг/кг (1,0 масс.% водный раствор), что превышает эффект метилурацила, 2,7 – в дозе 7 мг/кг (гель для приема внутрь 0,4 масс.%, гелеобразователь – хитозан), подтверждается данными гистологических исследований и превосходит эффективность висмута трикалия дицитрата.
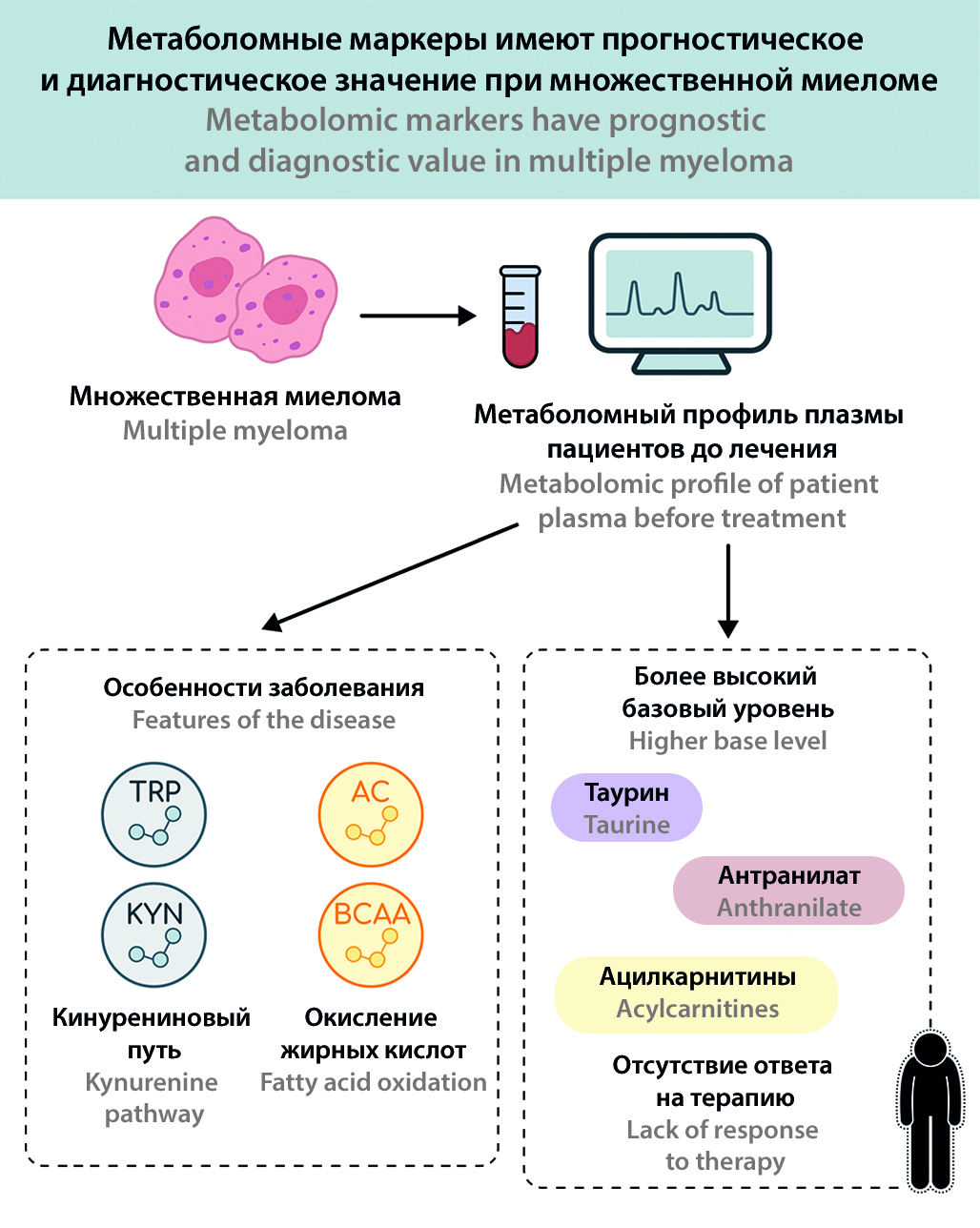
Введение. Множественная миелома (ММ) представляет собой злокачественное заболевание плазматических клеток, характеризующееся выраженной гетерогенностью клинического течения и вариабельностью ответа на лечение. Метаболомный анализ, отражающий совокупность малых молекул в биологических жидкостях, открывает новые возможности для поиска диагностических и прогностических биомаркеров.
Цель. Оценить метаболомные профили пациентов с ММ и выявить метаболические маркеры, ассоциированные с эффективностью полихимиотерапии.
Материалы и методы. Исследование проводилось с сентября 2022 года по май 2025 года на базе кафедры госпитальной терапии № 1 Сеченовского Университета. Проведен целевой анализ метаболитов плазмы крови 29 пациентов с ММ до начала лечения и 30 здоровых добровольцев (контроль). Пациенты были разделены на группы с ответом и без ответа на основании результатов проведения трех курсов терапии первой линии по протоколу VCD.
Результаты и обсуждение. Обнаружены значительные различия в метаболомных профилях пациентов с ММ по сравнению с контрольной группой. У больных ММ отмечен повышенный катаболизм триптофана через кинурениновый путь (~41 % увеличение соотношения кинуренин/триптофан, ~80 % снижение уровня серотонина), изменения в метаболитах цикла мочевины и оксида азота (~28 % снижение аргинина, ~5,3-кратное увеличение асимметричного диметиларгинина), а также аминокислотный дисбаланс (снижение уровня серина, аспартата, BCAA) и значительное увеличение общего количества ацилкарнитинов (в ~1,4 раза выше, чем в контроле). Исходный метаболический профиль также отличался у пациентов с различными результатами лечения: до лечения у пациентов, у которых впоследствии наблюдался клинический ответ, были снижены уровни некоторых ацилкарнитинов и продуктов распада триптофана (например, антраниловой кислоты), в то время как у пациентов без ответа были снижены уровни 5-гидрокситриптофана, индол-3-молочной кислоты и гистидина.
Заключение. Метаболомный анализ выявил характерные метаболические изменения при ММ, отражающие активацию иммунометаболических путей (триптофан-кинурениновый путь, метаболизм аргинина) и нарушение регуляции энергии и аминокислот. Полученные результаты указывают на потенциальную прогностическую значимость метаболитов: ряд биомаркеров (например, производные триптофана, ацилкарнитины) может быть связан с чувствительностью к химиотерапии. Полученные данные открывают перспективы для дальнейших исследований метаболических подходов в мониторинге и терапии ММ.
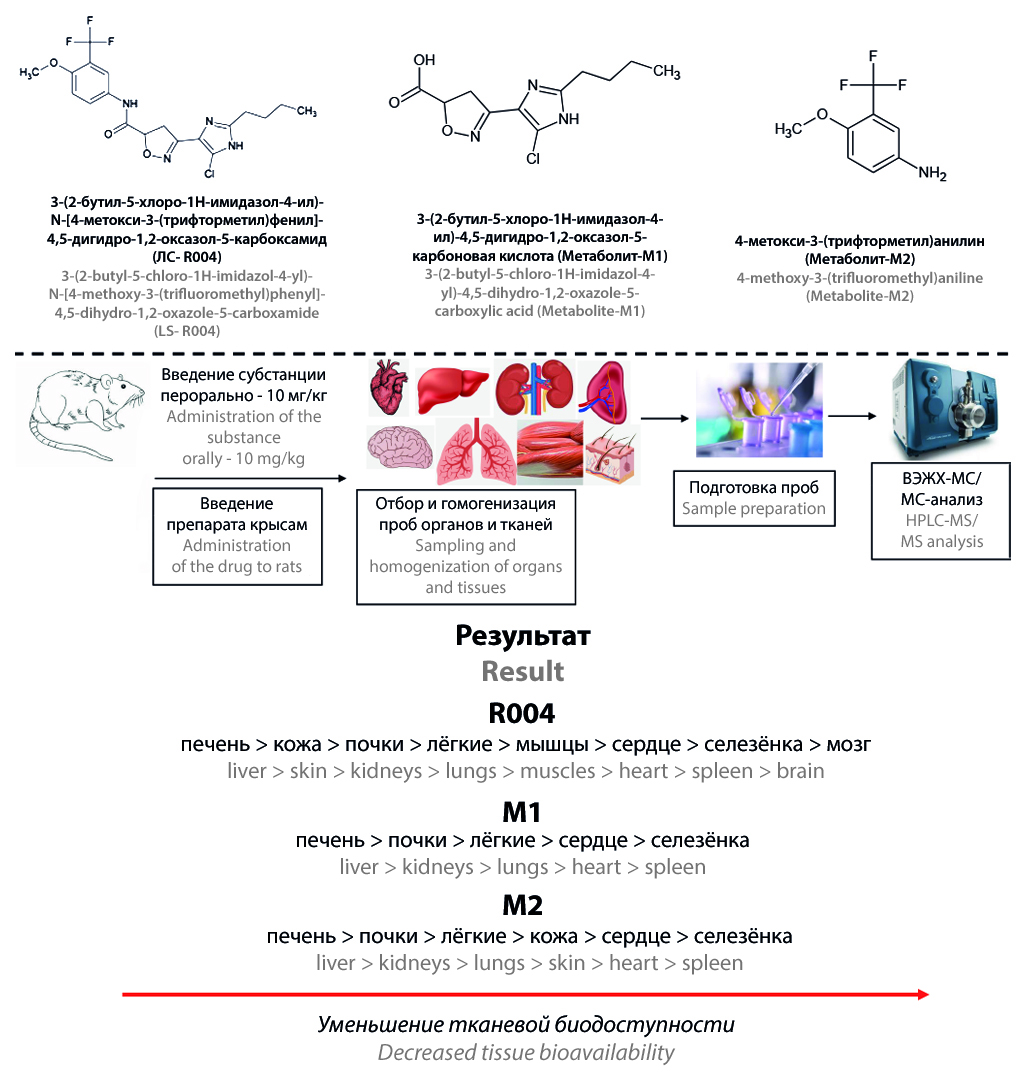
Введение. Новое фармакологически активное вещество 3-(2-бутил-5-хлоро-1Н-имидазол-4-ил)-N-[4-метокси-3-(трифторметил)фенил]-4,5-дигидро-1,2-оксазол-5-карбоксамид (R004) является новым ингибитором PAR-2-рецепторов для терапии ревматоидного артрита. Данное соединение находится на этапе доклинического исследования. Изучение распределения R004 и его метаболитов по органам и тканям ранее не проводилось.
Цель. Изучение распределения R004 и его метаболитов по органам и тканям крыс после однократного перорального введения субстанции.
Материалы и методы. Исследование проводилось на 50 крысах-самцах линии Wistar. Субстанция R004 вводилась перорально в терапевтической дозировке 10 мг/кг. Отбор образцов печени, почек, сердца, легких, селезенки, мозга, кожи и мышц проводился через 1, 2, 3, 4, 6, 8, 10, 12, 16, 24 ч после введения лекарственного средства. Органы немедленно гомогенизировались с использованием ацетонитрила и замораживались. Дальнейшую пробоподготовку осуществляли путем добавления к гомогенатам ацетонитрильного раствора внутренних стандартов. Количественное определение R004 и его метаболитов 3-(2-бутил-5-хлоро-1Н-имидазол-4-ил)-4,5-дигидро-1,2-оксазол-5-карбоновой кислоты (М1) и 4-метокси-3-(трифторметил)анилина (M2) проводилось методом ВЭЖХ-МС/МС.
Результаты и обсуждение. Биоаналитическая методика количественного определения R004, M1 и M2 в органах и тканях была успешно валидирована в аналитических диапазонах 2–2000 нг/г для R004 и 1–1000 нг/г для М1 и М2. R004 распределяется по всем изучаемым биологическим объектам. Тканевая биодоступность R004 уменьшается в следующей последовательности: печень > кожа > почки > легкие > мышцы > сердце > селезенка > мозг. М1 и М2 в больших количествах обнаружены в печени и почках. В сердце, легкие и селезенку метаболиты проникали в значительно меньших концентрациях. В тканях кожи идентифицирован только М2. В мозге и мышцах М1 и М2 не обнаружены.
Заключение. Валидированная биоаналитическая методика была успешно использована при изучении распределения субстанции R004. Действующее вещество хорошо распределяется по всем изучаемым органам и имеет высокую тканевую биодоступность. Наибольшие концентрации метаболитов зафиксированы в печени и почках.
Введение. Интермиттирующая гипоксия (ИГ) способствует свободнорадикальному окислению кислорода, что может предшествовать многим заболеваниям. Снижение физической активности, ишемические процессы в органах и нарушения на клеточном уровне, могут быть следствием прерывистой гипоксии. Актуальным является поиск потенциальных ЛС для коррекции данного процесса.
Цель. Сравнительное изучение эффективности активного метаболита этилметилгидроксипиридина сукцината (ЭМГПС) – этилметилсульфапиридина (ЭМСП), с нативной молекулой на модели ИГ у мышей.
Материалы и методы. Введение исследуемых объектов осуществляли внутрибрюшинно в течение 14 дней – ЭМСП вводили в дозе 85 мг/кг, Мексидол® – в дозе 100 мг/кг. Длительную интермиттирующую гипоксию воспроизводили путем помещения животных в мембранный гипоксикатор. Устанавлен следующий режим в течение 14 дней: 6 % – содержание кислорода в гипоксической камере, продолжительность – 6 часов. Оценивали влияние препарата на динамическую нагрузку (тест «сила хвата»), параметры дыхания (показатели плетизмографа), поведенческие и когнитивные показатели (тесты «открытое поле» и «приподнятый крестообразный лабиринт»), частоту сердечных сокращений и насыщение венозной крови кислородом, а также изучали потенциальный механизм действия методом ПЦР realtime.
Результаты и обсуждение. Было выявлено, что ЭМСП проявлял эффективность по параметрам плетизмографии, в частности помогал адаптироваться организму к хроническому гипоксическому воздействию, что выражалось в значимых отличиях по показателям вдоха и выдоха от контрольной группы. Исследование поведенческих и когнитивных состояний выявили наличие тревожности, снижение исследовательской активности и увеличение подвижности животных во всех группах. У животных, которым вводили ЭМСП и Мексидол® данные параметры были менее выражены, чем в контрольной группе. Была отмечена тенденция к увеличению экспрессии гена, влияющего на комплекс убихинол-цитохром с-редуктазы, являющимся частью митохондриального дыхания.
Заключение. Согласно результатам исследования, ЭМСП продемонстрировал защитные свойства, сравнимые с нативной молекулой ЭМГПС. Также была выявлена тенденция к усилению стимуляции гена UQCRC2 на фоне введения ЭМСП по сравнению с ЭМГПС.

Введение. Хроническая сердечная недостаточность (ХСН) часто приводит к прогрессирующей дисфункции почек, однако стратегии фармакологической защиты, направленные на коррекцию метаболических и окислительных нарушений в почечной ткани, остаются недостаточно разработанными. В связи с этим представляется актуальным изучение влияния новых соединений, таких как производные малоновой кислоты, в том числе 4-[(3-этокси-3-оксопропаноил)амино]бензойная кислота (этмабен), на экспрессию генов, кодирующих ключевые ферменты метаболических и антиоксидантных путей в почках при ХСН. Ранее для этмабена было выявлено более выраженное нефропротективное действие, нежели для другого малоната – малобена.
Цель. Оценить влияние 4-[(3-этокси-3-оксопропаноил)амино]бензойной кислоты на экспрессию генов, регулирующих ферментативные пути в почках крыс с экспериментальной ХСН.
Материалы и методы. Исследование проведено на 30 аутбредных белых самцах крыс (300–350 г), содержавшихся в стандартных условиях (12-часовой световой режим, температура 22 ± 2 °C, влажность 50–60 %, доступ к корму и воде ad libitum). Животные были разделены на три группы. Группа 1 (Контроль–) состояла из здоровых животных (n = 10), которые получали очищенную воду (1 мл/кг/сутки, внутрижелудочно). Группа 2 (ХСН–) включала животных с хронической сердечной недостаточностью (ХСН, n = 10), получавших с 30-го дня после операции очищенную воду (1 мл/кг/сутки, внутрижелудочно). Группа 3 (ХСН+) состояла из животных с ХСН (n = 10), которым с 30-го дня после операции внутрижелудочно вводили этмабен в дозе 60 мг/кг/сутки. Модель ХСН воспроизводили лигированием левой коронарной артерии. Успешность модели подтверждали эхокардиографически (фракция выброса <40 %) на 30-й день после моделирования. Прооперированных животных рандомизировали методом случайного выбора. Этмабен или воду вводили ежедневно в течение 30 дней, начиная с 30-го дня после операции. На 61-й день животных эвтаназировали, ткани почек гомогенизировали в реактиве «Лира» (ООО «Биолабмикс», Россия). РНК экстрагировали с использованием хлороформа, изопропанола и NaOAc, очищали с помощью LiCl. Концентрацию РНК определяли на Nanophotometer N60, качество – гель-электрофорезом. Обратную транскрипцию проводили из 1 мкг РНК (MMLV-RT-Kit, Eurogen). ПЦР в реальном времени выполняли на QuantStudio 5 с SYBR Green (Eurogen), анализируя гены Gpx1, Nrf2, Nox1, Glud1, Hes1, mTOR, Txrnd1, HIF1a, Cpt1b, B2M (референс). Статистическую обработку проводили в R-Studio (версия 4.3.3) с использованием пакета RQdeltaCT. Применяли однофакторный дисперсионный анализ ANOVA, тест Краскера – Уоллиса, апостериорные тесты Тьюки и Данна, а также анализ отношения шансов и логистическую регрессию. Для анализа экспрессии генов использовали метод 2–ddCt с нормализацией относительно референсного гена B2M.
Результаты и обсуждение. Выявлено, что ХСН приводит к значительной активации транскрипционного фактора Nrf2, однако уровень экспрессии его целевого гена GPx1 оставался неизменным как при ХСН, так и при лечении этмабеном, что указывает на нарушение функционирования данного сигнального пути. Применение 4-[(3-этокси-3-оксопропаноил) амино]бензойной кислоты вызывало статистически значимое увеличение экспрессии гена Cpt1b (p < 0,05), что свидетельствует о смещении клеточного метаболизма в сторону β-окисления жирных кислот. Кроме того, зафиксированы достоверное подавление экспрессии прооксидантного фермента Nox1 и активация гена тиоредоксинредуктазы-1 (Txnrd1) (p < 0,05) в группах, получавших терапию. Сигнальный путь Notch активировался под воздействием этмабена, что проявлялось в повышении экспрессии гена Hes1 (p < 0,05), тогда как значимого влияния на экспрессию гена mTOR обнаружено не было.
Заключение. Этмабен функционирует как метаболический модулятор и редокс-регулятор. Его действие не связано с прямой антиоксидантной активностью, а обусловлено активацией адаптивных механизмов: уменьшением потребности в антиоксидантной защите за счет подавления источников реактивных кислородных форм (Nox1), активацией ключевого регулятора антиоксидантного ответа (Nrf2), а также изменением энергетического метаболизма через индукцию Cpt1b и Glud1.

Введение. Малобен – новый лекарственный препарат для лечения заболеваний печени с не изученной ранее фармакокинетикой у человека.
Цель. Определение фармакокинетических параметров препарата «Малобен», таблетки (ФГБОУ ВО СПХФУ Минздрава России), при однократном и многократном применении у здоровых добровольцев в рамках клинического исследования (КИ) I фазы.
Материалы и методы. КИ I фазы проводилось в два этапа. В первом этапе участвовали 24 добровольца, разделенных на 3 когорты по 8 человек: добровольцы когорты 1 получали по 60 мг малобена однократно, когорты 2 – по 120 мг однократно, когорты 3 – по 180 мг однократно. Во втором этапе участвовали добровольцы, завершившие участие в предыдущем этапе: добровольцы когорты 4 получали суточную дозу 60 мг, когорты 5 – суточную дозу 120 мг, когорты 6 – суточную дозу 180 мг. Для определения концентраций малобена в плазме крови добровольцев использовался метод высокоэффективной жидкостной хроматографии с тандемным масс-спектрометрическим детектированием (ВЭЖХ-МС/МС). На основании полученных в ходе аналитического этапа концентраций малобена в плазме крови добровольцев после однократного и многократного перорального приема малобена были изучены его фармакокинетические параметры.
Результаты и обсуждение. Фармакокинетические параметры малобена оценивались в 6 когортах по 8 добровольцев. Средние значения Cmax составили 18,09 ± 8,06, 41,36 ± 5,63 и 51,81 ± 11,05 нг/мл при однократном дозировании в когортах 1–3 и 37,93 ± 20,98, 70,83 ± 37,37 и 78,98 ± 37,03 нг/мл при многократном дозировании в когортах 4–6. Средние значения AUC(0–t) составили 348,59 ± 200,65, 938,32 ± 344,95 и 1177,13 ± 221,81 нг · ч/л при однократном дозировании в когортах 1–3 и 3142,22 ± 2091,08, 5714,73 ± 2482,56 и 7799,02 ± 3829,67 нг · ч/л при многократном дозировании в когортах 4–6. Средние значения AUC(0–∞) составили 623,05 ± 390,08, 1171,68 ± 471,89 и 1666,93 ± 596,25 нг · ч/л при однократном дозировании в когортах 1–3 и 3228,41 ± 2141,08, 5789,32 ± 2539,34 и 8970,72 ± 5143,42 нг · ч/л при многократном дозировании в когортах 4–6. Установлена пропорциональная зависимость фармакокинетических параметров Cmax, AUC(0–t) и AUC(0–∞) от величины дозы (линейность ФК) при однократном дозировании, AUC(0–t) и AUC(0–∞) – при многократном дозировании.
Заключение. Впервые была изучена ФК малобена при различных режимах дозирования у добровольцев, а также изучены закономерности изменения фармакокинетических параметров.
РЕГУЛЯТОРНЫЕ ВОПРОСЫ

Введение. Обеспечение качества исследовательских работ является ключевым фактором научных открытий, а также появления на рынке инновационных высокомаржинальных продуктов. В связи с особенностью научно-исследовательской деятельности единые правила по организации и проведению исследований, удовлетворяющие потребностям как научных групп, так и бизнеса, заинтересованного в коммерциализации разработок, в том числе разработок, связанных с лекарственными средствами, в данный момент отсутствуют, а индивидуальные подходы только формируются. Совокупность сложившихся подходов и концепций, согласно существующей системе надлежащих практик (GxP), получила название «Надлежащая исследовательская практика», или GRP.
Цель. Обобщение существующих мировых трендов в области надлежащей исследовательской практики и описание реализации принципов GRP на примере системы менеджмента качества лабораторного комплекса Университета «Сириус».
Материалы и методы. Методы реализации принципов GRP в лабораторном комплексе базируются на требованиях ISO 9001 и рекомендациях ICH в таких областях, как управление документами и записями, управление персоналом и обучением, управление аудитами и инспекциями, управление рисками, анализ и оценка результативности и эффективности деятельности.
Результаты и обсуждение. Проанализированы текущие принципы GRP, сформированные в рамках национальных научных сообществ, отдельных университетов и государственных программ. На базе лабораторного комплекса Университета «Сириус» рассмотрены возможности и примеры комплексной имплементации GRP в такие области системы менеджмента качества, как управление документами и записями, управление персоналом и обучением, управление аудитами и инспекциями, управление рисками, проведение периодического анализа и оценки результативности и эффективности деятельности.
Заключение. Описанный в статье подход к проведению научно-исследовательских работ, основанный на комплексной имплементации GRP, является инструментом обеспечения качества исследований, необходимым как для сугубо научной деятельности, так и для успешного научно-прикладного взаимодействия между академической средой и фармацевтическими компаниями с целью ускорения вывода на рынок новых лекарственных средств и удовлетворения запроса пациентов.

Введение. Вывод на рынок лекарственных средств для медицинского применения – чрезвычайно дорогостоящий и ресурсоемкий процесс, связанный с длительной разработкой, высокими рисками, гарантией качества конечного продукта, безопасностью и эффективностью, при этом ограниченный жесткими регуляторными требованиями. Ключевыми целями развития фармацевтической промышленности являются содействие условиям гарантии безопасности Российской Федерации в сфере лекарственного обеспечения населения и лекарственной доступности во всех сегментах, обеспечение передового уровня научно-технического и технологического развития фармацевтической промышленности, создание экспортно ориентированного потенциала, наличие компетенций в исследованиях и разработках, производстве полного цикла, внедрении в клиническую практику и экспорте лекарственных средств. Развитие фармацевтической и медицинской промышленности Российской Федерации предполагает не только последовательно проводимую политику импортозамещения, но и задачи по реализации экспортного потенциала для обеспечения комплексного поддержания стабильности национального фармацевтического рынка.
Текст. В статье продемонстрированы данные по экспорту российских лекарственных средств, предложены возможные способы и инструменты для снижения барьеров при выходе на зарубежные рынки и поддержки экспортной деятельности отечественных производителей.
Заключение. Основные направления реализации Стратегии развития фармацевтической промышленности Российской Федерации на период до 2030 года предусматривают поддержку экспорта российской фармацевтической продукции. Применение системы оценки уровней готовности экспортного потенциала позволит использовать ее в качестве инструмента для снижения барьеров при выходе на зарубежные рынки и усилить позиции российских производителей, в том числе обеспечить гармонизацию регулирования с наилучшими мировыми практиками.
ЮБИЛЕЙ

Новости отрасли
2026-01-29
Прием заявок в номинацию «Проект года» конкурса «XXVI Платиновая Унция» завершится 31 января
 | 31 января завершится прием заявок в номинацию «Проект года» конкурса «XXVI Платиновая Унция» |
2026-01-27
33-й Российский фармацевтический форум: время планировать участие
 | Ведущая отраслевая площадка для профессионалов фармрынка приглашает всех участников рынка присоединиться к крупнейшему событию фармацевтической отрасли России. |
2026-01-26
Российская вакцина против ВПЧ одобрена для применения у детей
 | Министерство здравоохранения Российской Федерации одобрило применение первой отечественной вакцины против вируса папилломы человека (ВПЧ) «Цегардекс» у детей в возрасте от 9 до 17 лет. Соответствующие изменения внесены в документы регистрационного досье вакцины. Актуализированная инструкция по медицинскому применению вакцины «Цегаредкс» опубликована 23 января в Государственном реестре лекарственных средств (ГРЛС). |
| Ещё новости |

ISSN 2658-5049 (Online)