



ОТ РЕДАКЦИИ

15 июня 2024 года исполняется 80 лет со дня рождения заведующего кафедрой фармацевтической химии и фармацевтической технологии фармацевтического факультета Воронежского государственного университета профессора, доктора фармацевтических наук, заслуженного работника высшей школы Российской Федерации Алексея Ивановича Сливкина.
Во второй части статьи представлена структура Аптекарского приказа в момент его окончательного становления. Обращено внимание на кадры, служившие в Приказе. Перечислены руководители Приказа с момента его возникновения и до его окончательной реорганизации, а также должности основного и вспомогательного персонала. Разобраны основные функции Аптекарского приказа, которые традиционно разделяются на дворцовые и государственные. К первой группе относят лечебную, ветеринарную, заготовительную, садоводческую, экспертную и производственную функции. К общегосударственным относятся медико-полицейская, военно-медицинская, образовательная и управленческая функции. Особый интерес представляет функция Аптекарского приказа по сбору, хранению и изучению научных сочинений по медицине.

Фармацевтические препараты и лекарственные средства играют важную роль в поддержании здоровья человека. Однако, как и любые другие продукты, они могут содержать потенциально опасные вещества, так называемые генотоксичные примеси, которые могут нанести вред здоровью человека при их употреблении, поэтому контроль качества фармацевтической продукции имеет особое значение. Генотоксичность — это способность вещества оказывать необратимое действие на структуру и функции ДНК в клетках, тем самым вызывая гибель ДНК и ошибки в ее репликации, мутации и хромосомные аберрации. Одними из таких веществ являются N-нитрозамины, которые образуются в результате взаимодействия вторичных аминов с нитритами или нитратами. Группа компаний Лабконцепт – официальный дистрибьютор ведущих производителей аналитического и общелабораторного оборудования. Лабконцепт предлагает комплексные решения для оснащения лабораторий разных направленностей, в том числе и для фармацевтической отрасли. В данной статье мы рассмотрим возможность определения таких примесей с использованием газового хромато-масс-спектрометра с тройным квадруполем EXPEC G-Chrom MS марки Expec на примере N-нитрозаминов.
МЕРОПРИЯТИЯ

IX Всероссийская GMP-конференция с международным участием прошла 21–23 августа в столице Республики Башкортостан – Уфе. Девиз этого года – «GMP – непрерывное развитие».
ПОИСК И РАЗРАБОТКА НОВЫХ ЛЕКАРСТВЕННЫХ СРЕДСТВ

Введение. Представители семейства вересковых довольно распространены на территории России и перспективны для создания новых лекарственных средств растительного происхождения. В то же время официнальными из них являются только 4 вида. Перспективно исследование биологически активных веществ и фармакологической активности Andromeda polifolia L., Chamaеdaphne calyculata (L.) Moench, Ledum palustre L., Empetrum nigrum L., обладающих богатыми ресурсными запасами.
Цель. Сравнительное исследование компонентного состава суммарных фракций флавоноидов A. polifolia, C. calyculata, L. palustre, E. nigrum и изучение их влияния на NO-стимулирующую активность перитонеальных макрофагов.
Материалы и методы. Измельченную надземную часть (облиственные побеги) предварительно депигментировали хлороформом, обрабатывали 70%-м водным ацетоном, ацетон удаляли. Флавоноиды экстрагировали этилацетатом из водной фазы. Идентификацию флавоноидов проводили методом ВЭЖХ (хроматограф UltiMate 3000) по совпадению времен удерживания и спектральных характеристик, расчет содержания – методом простой нормировки. Влияние образцов на продукцию оксида азота изучали на макрофагах мышей линии C57BL/6. Контроль эндотоксина в образцах осуществляли с помощью ЛАЛ-теста и инкубирования клеток в присутствии полимиксина B.
Результаты и обсуждение. Исследован компонентный состав фракций флавоноидов A. polifolia, C. calyculata, L. palustre, E. nigrum. В побегах C. calyculata обнаружены 8 фенольных соединений, в том числе изокверцитрин, гербацетин, нарингенин и нарингин – впервые для данного вида. В побегах A. polifolia выявлено 5 соединений, в том числе изокверцитрин и гербацетин – впервые для данного вида. В побегах L. palustre и E. nigrum идентифицированы 5 и 4 соединения соответственно, при этом во всех образцах преобладающими являются гликозиды кверцетина: изокверцитрин, гиперозид и рутин. Суммарная фракция флавоноидов C. calyculata в дозах 1, 5, 10 мкг/мл ингибирует продукцию оксида азота макрофагами на 30 %, а флавоноиды E. nigrum в дозах 100 и 200 мкг/мл, напротив, усиливают продукцию нитритов макрофагами на 33 и 37 % соответственно.
Заключение. Проведено сравнительное исследование компонентного состава суммарных фракций флавоноидов A. polifolia, C. calyculata, L. palustre, E. nigrum, которые способны активировать как М1-, так и М2-поляризацию перитонеальных макрофагов мышей, что требует дальнейшего углубленного изучения. Перспективными для дальнейшего изучения являются флавоноиды C. сalyculata и E. nigrum.
ФАРМАЦЕВТИЧЕСКАЯ ТЕХНОЛОГИЯ
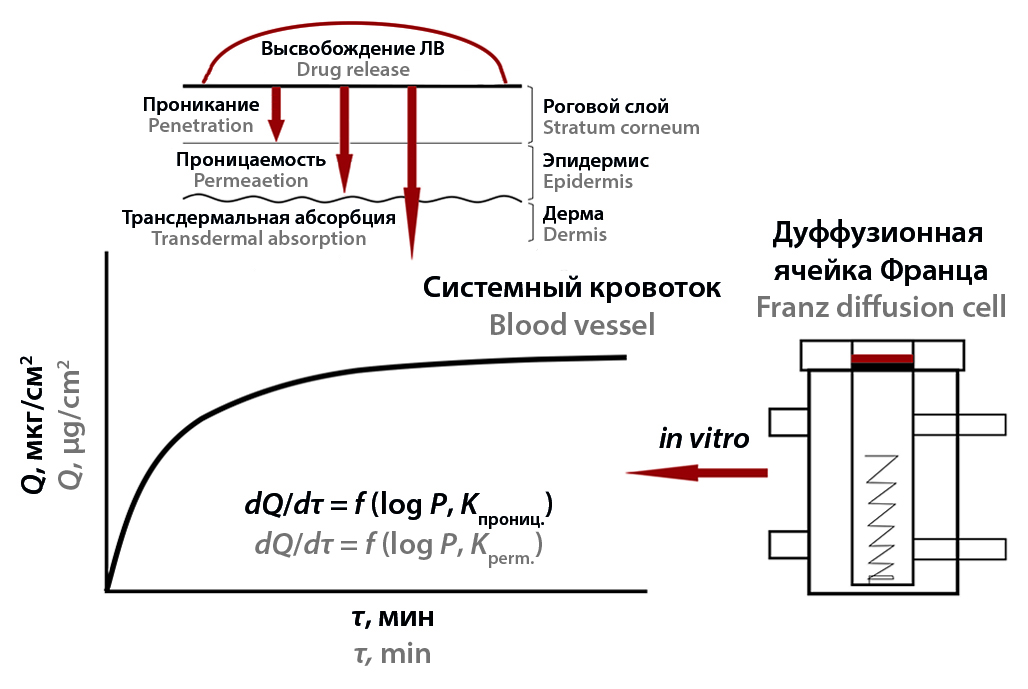
Введение. В обзоре обсуждаются основные концепции высвобождения лекарственных веществ (ЛВ) из лекарственных форм (ЛФ) и кинетическое моделирование этого процесса по профилю растворения с использованием вертикальной диффузионной ячейки Франца.
Текст. Высвобождение ЛВ из ЛФ (мази, гели, трансдермальные пластыри и полимерные пленки) обычно описывают как процесс растворения ЛВ в биологической системе. Формально этот процесс в соответствии с фармакопейными методами оценивают, используя различные тесты на растворимость. Полученные на основании этих тестов экспериментальные данные обычно выполняют также и прогностическую функцию по отношению к прониканию ЛВ через кожный барьер и проницаемости среды. Наиболее часто для оценки высвобождения ЛВ и прогнозирования проницаемости используют вертикальные диффузионные ячейки Франца, выбирая тип системы диффузионных ячеек и тип мембраны, которые необходимы для высвобождения конкретного ЛВ. Теоретические аспекты высвобождения базируются на теории массопереноса веществ из полимерной матрицы в систему, имитирующую биологическую среду. Высвобождение ЛВ может осуществляться через механизмы пассивной диффузии по Фику и «нефиковской» диффузии, десорбцию ЛВ с внутренней стороны мембраны, а также по другим механизмам. Высвобождение ЛВ определяется как его липофильностью, природой мембраны, так и различными физико-химическими параметрами ЛВ. Одной из корреляционных характеристик массопереноса является оценка коэффициента проницаемости для конкретной, имитирующей кожу мембраны, описывающего скорость проникания вещества на единицу концентрации в единицах «расстояние/время». Примером корреляционных соотношений «структура – проницаемость» (QSPeR или QSPR) являются уравнения, связывающие константу проницаемости и липофильность с молекулярной массой ЛВ. В работе рассмотрены статистические методы анализа данных (MANOVA, ANOVA) и модельно-зависимые методы (нулевой порядок, первый порядок, модель Хигучи, модель Корсмейера – Пеппаса, модель Хиксона – Кроуэлла и др.). Идеальная доставка недеградируемых и недезагрегируемых ЛВ, как правило, описывает модель высвобождения реакции нулевого порядка. Для водорастворимых ЛВ из пористой матрицы более характерны модели реакции первого порядка. Наиболее часто для описания процесса высвобождения из гелей и дермальных пленок и пластырей используют кинетические модели дробных степенных функций в виде зависимости потока от времени в степени τ1/2 (модель Хигучи) или τ1/3 (модель Хиксона – Кроуэлла). Модель Корсмейера – Пеппаса позволяет оценить механизм массопереноса с диффузией по Фику или по другому процессу.
Заключение. Математическое моделирование кинетики высвобождения ЛВ из мягких ЛФ является важным элементом для разработки и оптимизации их составов. Исследование высвобождения ЛВ из мягких ЛФ, в том числе ТТС и полимерных пленок, так же как и из твердых ЛФ, основано на установлении корреляционных соотношений кинетики профиля высвобождения и растворения. Основными моделями высвобождения независимо от ЛФ остаются модели: нулевого порядка, первого порядка, Корсмейера – Пеппаса, Хигучи, Хиксона – Кроуэлла, эмпирические или полуэмпирические константы которых существенно различаются в зависимости от ЛФ и механизма высвобождения (диффузия по Фику или другой механизм массопереноса ЛВ). Корреляционные соотношения QSPeR или QSPR, использующие коэффициенты проницаемости (Кпрониц.), диффузии и липофильности, позволяют получить информацию о массопереносе ЛВ через кожу.
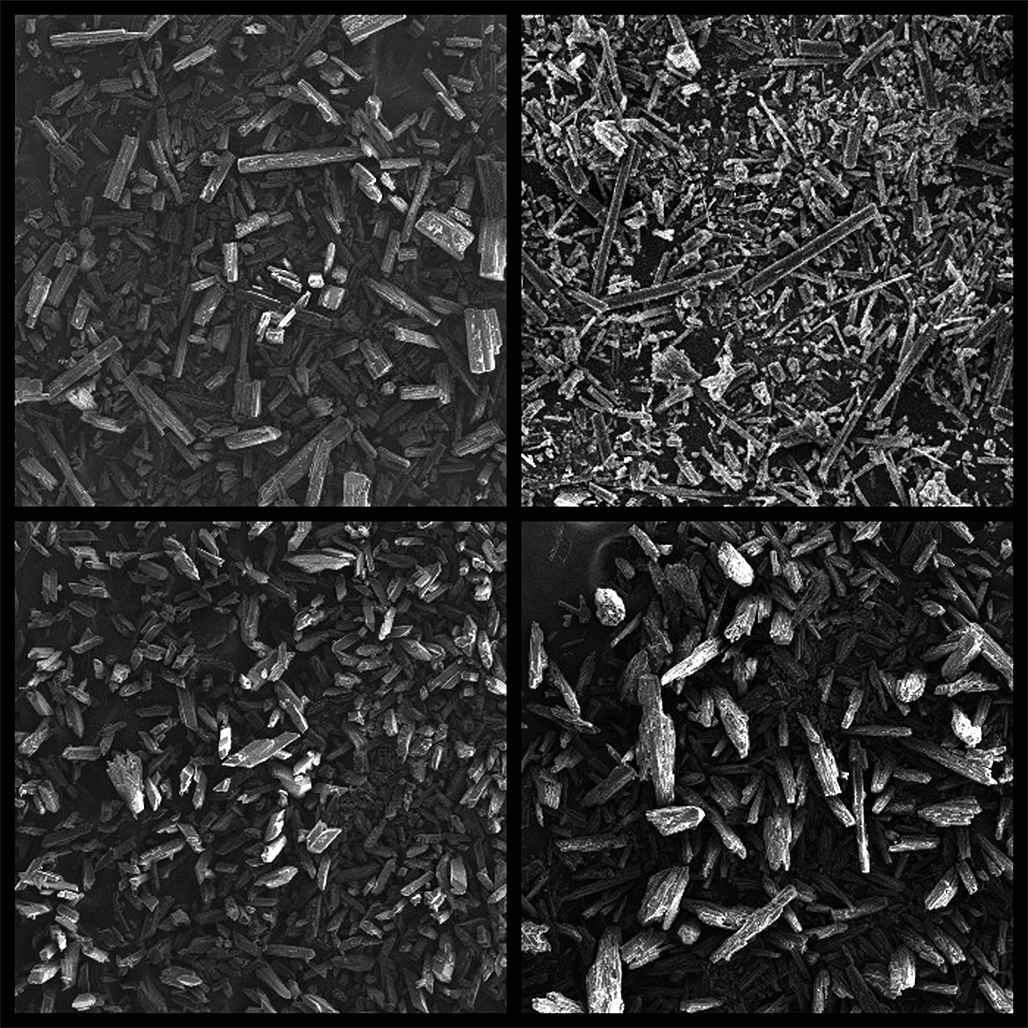
Введение. В современном мире существует множество фармацевтических субстанций, отличающихся по структуре. При разработке готовых лекарственных форм (ГЛФ) необходимо учитывать множество факторов, таких как физико-химические и технологические свойства субстанций и вспомогательных компонентов, технология производства и др. Данная работа посвящена изучению этих факторов на примере субстанции, имеющей анизодиаметрическую форму кристаллов. Ребамипид был выбран как пример такой субстанции с плохими технологическими характеристиками для исследования и подбора технологии.
Цель. Исследование подходов к изучению субстанции с анизодиаметрической формой кристаллов на примере ребамипида для установления его физико-химических и технологических свойств с целью теоретического обоснования наилучшего способа получения массы для таблетирования.
Материалы и методы. В качестве материалов использовался ребамипид (N-(4-хлорбензоил)-3-(2-оксо-1,2-дигидрохинолин-4-ил)аланин) (экспериментальный образец). В качестве оборудования использовался тестер сыпучести ERWEKA GT (ERWEKA GmbH, Германия), тестер насыпной плотности SVM 122 (ERWEKA GmbH, Германия), вибросито CISA RP 200N (CISA Cedaceria Industrial S.L., Испания), порошковый дифрактометр D8 ADVANCE (Bruker Corporation, США), калориметр DSC 204 F1 (NETZSCH, Германия), электронный микроскоп Hitachi TM-100 (Hitachi, Япония).
Результаты и обсуждение. Была проведена оценка свойств субстанции ребамипида. Исследован полиморфизм субстанции, температура плавления, микроскопия и оценка технологических характеристик субстанции. Применение метода SeDeM позволило определить критические показатели субстанции, которые необходимо скорректировать.
Заключение. При исследовании субстанции ребамипида экспериментальным путем выяснено, что данное вещество обладает полиморфизмом, имеет высокую температуру плавления, иглообразную форму кристаллов, плохую сыпучесть и уплотняемость, что подтверждено применением метода SeDeM.
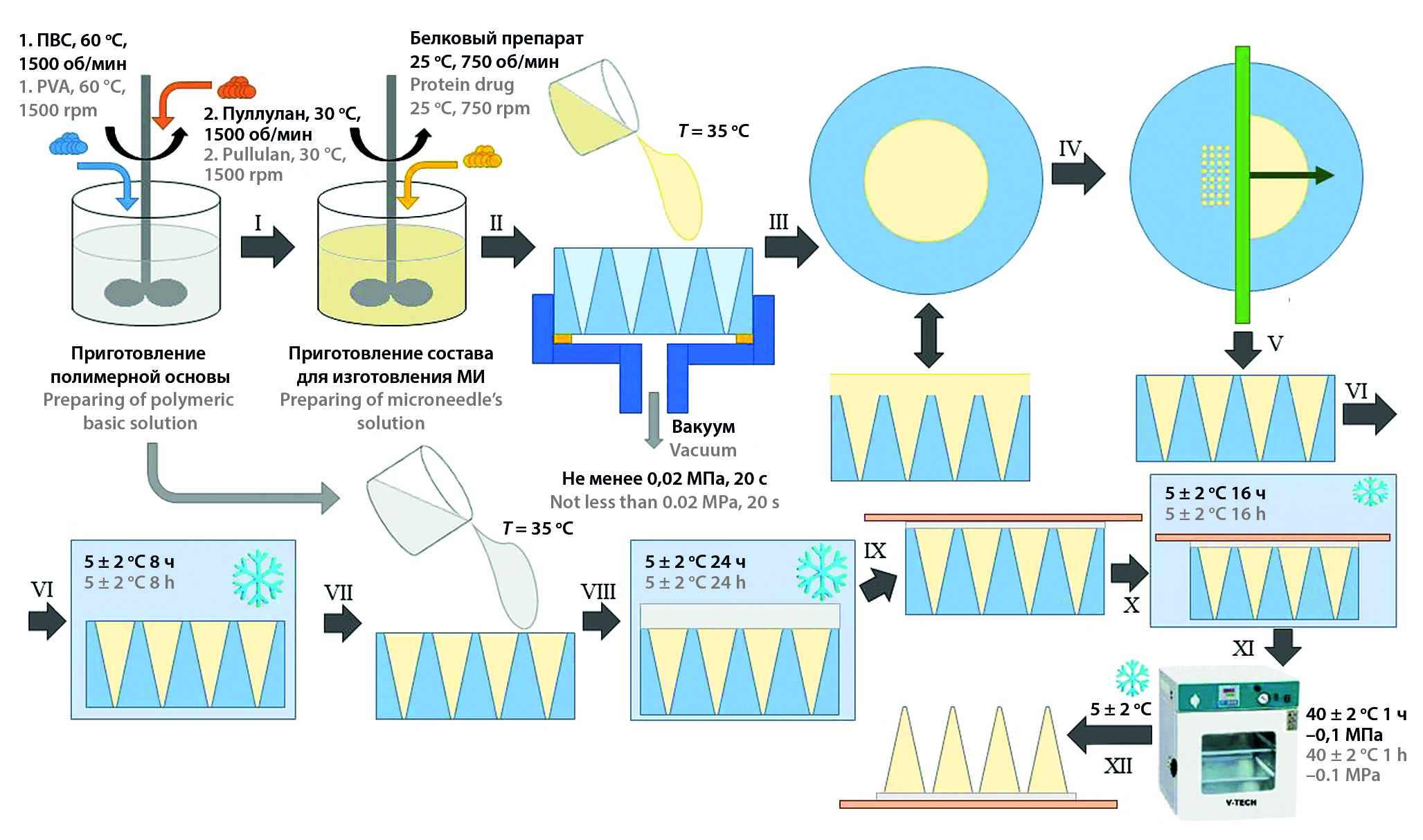
Введение. Растворяющиеся полимерные микроиглы являются перспективной системой доставки лекарственных препаратов, в частности вакцин. Однако до сих пор существуют проблемы при разработке оптимальной масштабируемой технологии их изготовления.
Цель. Разработать масштабируемую технологию изготовления полимерных растворяющихся микроигл, которая позволит максимально сохранить белковые препараты в производственном процессе.
Материалы и методы. Для изготовления микроигл использовался метод отлива из растворителя в мастер-формы из полиэтилентерефталата со сквозными микрополостями конической формы. В качестве материала микроигл использовался водный раствор, содержащий 20 % масс. об. пуллулана и 3 % масс. об. поливинилового спирта. В качестве модельного белка использовался препарат человеческого сывороточного альбумина.
Результаты и обсуждение. В ходе работы были подобраны оптимальные режимы заполнения мастер-формы и сушки микроигл, позволяющие максимально сохранить белковый препарат в составе микроигл в процессе производства.
Заключение. Разработанная технология изготовления полимерных растворяющихся микроигл может быть масштабирована, так как не содержит лимитирующих стадий производства, и использоваться для производства систем доставки вакцин.

Введение. Разработка бездефектных режимов нанесения пленочной оболочки является актуальной задачей при проведении трансфера технологии таблеток, покрытых пленочной оболочкой. Трансфер технологий – неотъемлемая стадия жизненного цикла любого лекарственного средства, допущенного к промышленному производству. Во время проведения отработки технологического процесса могут возникнуть отклонения, для устранения которых требуется вводить вспомогательные операции или менять цепочку технологического оборудования.
Цель. Оптимизировать процесс нанесения пленочного покрытия на таблетки ядра витаминно-минерального комплекса.
Материалы и методы. Объектами исследования были выбраны двояковыпуклые таблетки-ядра в форме лодочки витаминно-минерального комплекса, состоящего из витаминов С + Е + В1 и минералов. Для обеспечения требуемых технологических свойств использовались вспомогательные вещества: микрокристаллическая целлюлоза, кроскармеллоза натрия, крахмал картофельный, кальция стеарат, кремния диоксид коллоидный. В качестве пленкообразующей композиции использовалась готовая пленкообразующая смесь (Colorcon®) бежевого цвета Opadry® II 85F 270000. Нанесение пленочной оболочки осуществлялось в коатерах с перфорированным барабаном BG-80 (Pro-face, Китай) и BGK-150 (Zhejiang Canaan Technology Limited, Китай).
Результаты и обсуждение. Изучен процесс нанесения пленочного покрытия на таблетки-ядра витаминно-минерального комплекса. Анализ действующей технологической цепочки показал ряд недостатков: большое количество ручного труда, необходимость матирования барабана, длительность процесса, небольшую загрузку, торможение производственного цикла из-за отставания на стадии нанесения пленочной оболочки. Для решения обнаруженных проблем был разработан проект масштабирования технологии на стадии нанесения пленочной оболочки на таблетки-ядра витаминно-минерального комплекса. В ходе реализации процесса было закуплено и смонтировано на производстве новое оборудование. Отработка технологического процесса позволила кратно увеличить единовременную загрузку в коатер (в 2–3 раза), автоматизировать процесс за счет функции автоматической работы по заданным до начала технологического процесса параметрам, исключить матирование барабана из подготовительных работ. Качество таблеток, покрытых пленочной оболочкой, улучшилось, риск возникновения дефектов значительно снизился.
Заключение. В ходе исследования было доказано, что перенос процесса нанесения пленочного покрытия на другую единицу оборудования может улучшить качество получаемых таблеток, покрытых пленочной оболочкой, и оптимизировать технологический процесс. Кратное увеличение размера загрузки не осуществляется за то же самое время за счет изменения количества форсунок и внутреннего сечения шланга для нанесения материала пленочного покрытия.
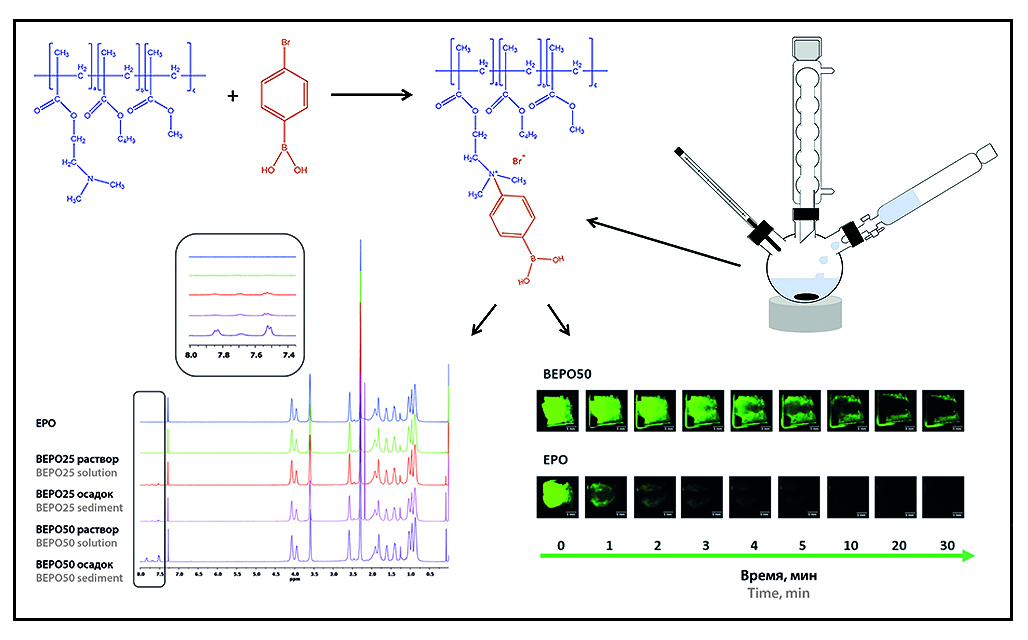
Введение. Большой интерес в области фармацевтической технологии проявляют к полимерам, обладающим мукоадгезивными свойствами, так как они увеличивают время пребывания лекарства на поверхности слизистой и тем самым повышают биодоступность препарата. Существуют различные мукоадгезивные системы доставки: таблетки, пленки, гели, суспензии на основе микро- и наночастиц и др. Способность к адгезии зависит от вспомогательных веществ полимерной природы, а именно от их химической структуры. Большую роль играют молекулярная масса, поверхностный заряд, гибкость полимерной цепи и наличие различных функциональных групп. Сополимеры под торговым наименованием Eudragit®, производимые немецким концерном Evonik Nutrition & Care GmbH, применяются в фармацевтической промышленности на протяжении нескольких десятилетий для получения пероральных лекарственных форм с контролируемым высвобождением. Eudragit® ЕРО (ЕРО), тройной сополимер на основе метакрилатных мономеров, обладает мукоадгезивными свойствами за счет наличия в своей структуре диметиламино групп. Предлагаемая его химическая модификация с помощью производного фенилбороновой кислоты, ввиду наличия гидроксильных групп в ее структуре, приводит к дополнительному взаимодействию с олигосахаридами муцина, обеспечивая усиление мукоадгезивных свойств Eudragit® ЕРО.
Цель. Синтез и исследование химически модифицированной формы Eudragit® ЕРО с применением 4-бромфенилбороновой кислоты с целью повышения мукоадгезивных свойств сополимера для использования в трансмукозальных системах доставки лекарств.
Материалы и методы. Синтез химически модифицированного Eudragit® ЕРО (ВЕРО) проводили в течение 24 ч при температуре 50 °С с последующей очисткой методом диализа с применением диализной мембраны (MMO = 12–14 кДa; Mеdicеll Intеrnаtionаl Ltd., Великобритания) в течение 7 дней и дальнейшим лиофильным высушиванием при –50 °С и 0,05 мБар с применением Hеtо Pоwеr Dry LL 3000 (Thеrmо Elеctrоn Cоrpоrаtiоn, США) в течение 5 дней. Подтверждение образования химически модифицированной формы ВЕРО проводили методами ИК-спектроскопии на приборе Nicоlеt iS5 (Thеrmо Fisher Sciеntific, США) и 1Н-ЯМР-спектроскопии на приборе DPX 400 МГц (Bruker, Германия). Термогравиметрический анализ (ТГА) и модулированная дифференциальная сканирующая калориметрия (мДСК) проводились на приборах Discоvеry ТGА™ и Discоvеry DSC™ (ТА Instrumеnts, США) соответсвенно. Изучение мукоадгезивных свойств проводилось по способности удерживания сополимера на изолированной слизистой носа овцы в течение 30 мин при температуре 37,0 ± 0,5 °С.
Результаты и обсуждение. Был получен ВЕРО со степенью замещения диметиламино групп фенилбороновой кислотой на 25 % (BEPO25) и 50 % (BEPO50). Выход BEPO25 составил 40,70 %, ВЕРО50 – 30,79 %. На ИК-спектрах ВЕРО появляются характеристические полосы в области 1605 см–1, которые указывают на присоединение фенилбороновой кислоты к ЕРО. На 1Н-ЯМР-спектрах ВЕРО наблюдается образование дополнительных пиков в диапазоне 7,8 и 7,5 ppm, которые отсутствуют на спектре ЕРО и указывают на наличие фенилбороновой кислоты. Согласно данным ТГА, полученные образцы модифицированного ЕРО характеризуются сопоставимой с исходным ЕРО термической стабильностью. Результаты анализа ДСК-термограмм свидетельствуют, что температуры стеклования (Tс) образцов ВЕРО несколько выше, чем исходного ЕРО, что может быть связано с уменьшением свободных диметиламино групп в составе терполимера. Образец ВЕРО50 удерживается на поверхности изолированной слизистой носа овцы в течение 30 мин, в то время как ЕРО смывается искусственной назальной жидкостью за 5 мин.
Заключение. Получение и исследование ВЕРО является перспективным направлением для дальнейшего использования в трансмукозальных системах доставки лекарств.
МЕТОДЫ АНАЛИЗА ЛЕКАРСТВЕННЫХ СРЕДСТВ
Введение. Определение содержания остаточных количеств пестицидов в лекарственном растительном сырье (ЛРС) и препаратах на его основе является важным этапом подтверждения безопасности на этапе контроля качества ЛРС. Для обработки лекарственных растений в процессе выращивания все более активно применяются фосфорорганические пестициды (ФОП). В литературе представлены сведения обнаружения ФОП в ЛРС, в том числе в количествах, превышающих пределы допустимого содержания, регламентированные нормативной документацией. Вместе с тем проблема распределения конкретных представителей ФОП в органах лекарственных растений (ЛР) и их персистентности остается недостаточно изученной.
Цель. Изучить распределение в различных частях ЛР и персистентность малатиона и диазинона в ноготках лекарственных, валериане лекарственной и крапиве двудомной.
Материалы и методы. В качестве модельных ЛР выбраны крапива двудомная, произрастающая повсеместно, ноготки лекарственные, исключительно культивируемые, и валериана лекарственная. Все растения разделили на три равные группы. Молодые побеги растений дважды обрабатывали средствами «Алиот», содержащим 570 г/л малатиона, и «Террадокс», содержащим 40 г/кг диазинона. Первую группу растений обрабатывали пестицидами согласно инструкции производителя, вторую группу – в количествах, в три раза превышающих рекомендуемую для внесения дозировку. Третья группа – контрольная – не подвергалась обработке пестицидами. Образцы различных частей растущих ЛР анализировались в определенные сроки методом ВЭЖХ-МС/МС по разработанной методике. Для изучения персистентности через 0,5 года осуществляли повторный анализ высушенных образцов, хранящихся без доступа солнечных лучей согласно требованиям Государственной фармакопеи РФ XV издания.
Результаты и обсуждение. Малатион и диазинон обнаруживались во всех анализируемых частях ноготков лекарственных, в листьях и корнях крапивы двудомной, в олиственных побегах и корневищах с корнями валерианы лекарственной. Наибольшее содержание пестицидов зафиксировано в подземных органах растений: при обработке согласно инструкции в корнях ноготков обнаружено 16,7 мг/кг сырья малатиона и 19,6 мг/кг диазинона, в корнях крапивы – 4,5 мг/кг малатиона и 4,1 мг/кг диазинона и в корневищах с корнями валерианы лекарственной – 1,7 мг/кг малатиона и 1,5 мг/кг диазинона, что превышает допустимые нормируемые значения. Содержание пестицидов в цветках ноготков лекарственных не превышает допустимые значения. Исследование персистентности данных ФОП свидетельствует о том, что малатион и диазинон сохраняются в тканях растений в течение длительного времени.
Заключение. По результатам исследования получена количественная характеристика распределения малатиона и диазинона в различных частях ноготков лекарственных, валерианы лекарственной и крапивы двудомной, а также скорость и степень деградации ФОП в течение полугодового периода.
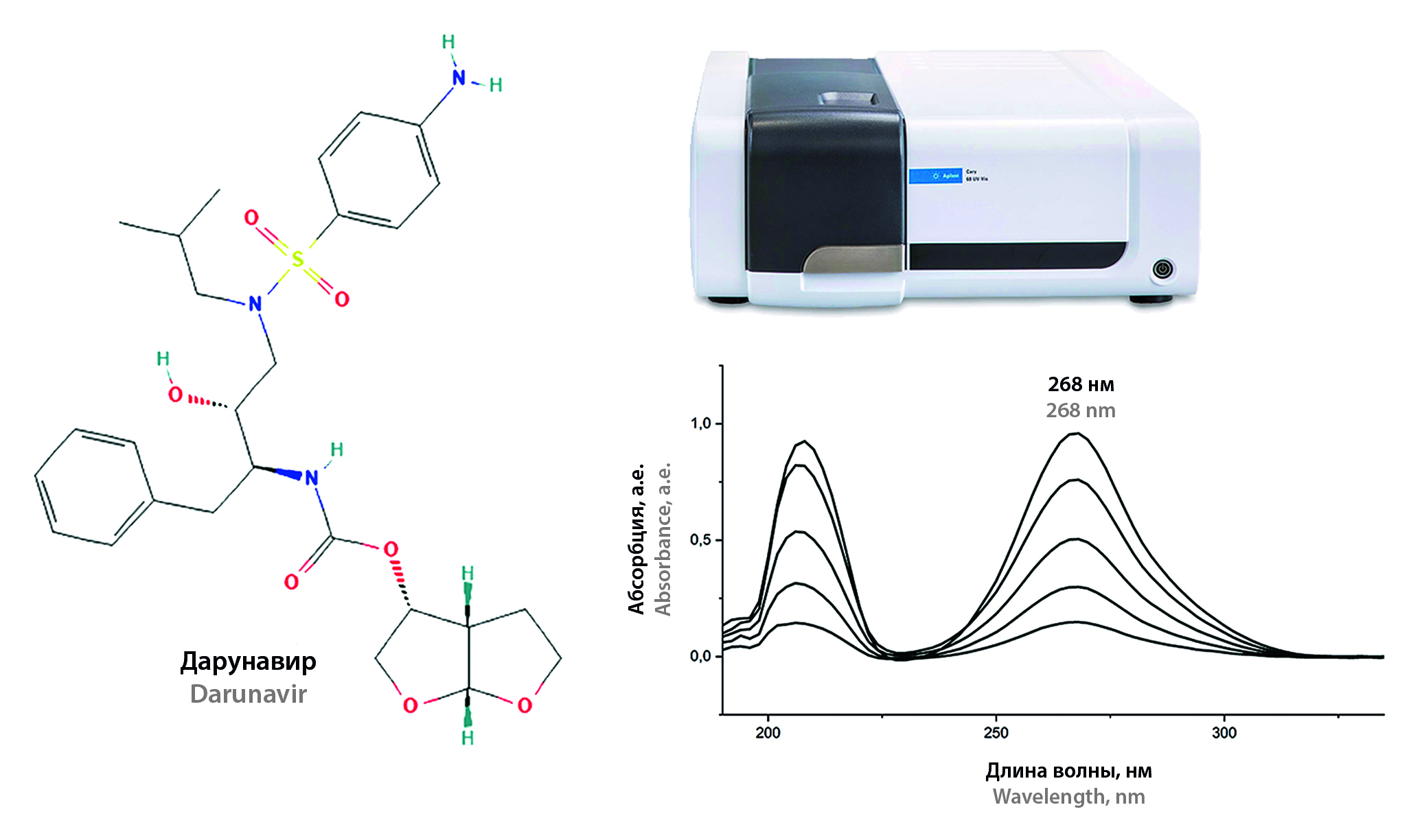
Введение. Дарунавир, являясь эффективным средством антиретровирусной терапии, широко применяется в клинической практике, в том числе для лечения педиатрических пациентов и беременных женщин, а также в качестве инструмента персонализированной терапии. При этом в производстве готовых лекарственных форм в настоящее время используется дарунавир как в виде кристаллического этанолата, так и в форме аморфной субстанции. В связи с этим существует потребность в разработке и совершенствовании методов количественного определения дарунавира. В качестве недорогой и эффективной альтернативы распространенным хроматографическим и титриметрическим методам может быть использовано спектрофотометрическое определение дарунавира в ультрафиолетовой области спектра (УФ-спектрофотометрия).
Цель. Разработать и валидировать методику количественного определения аморфного дарунавира в субстанции методом УФ-спектрофотометрии.
Материалы и методы. Для исследования использовали следующие субстанции и расходные материалы: субстанцию-порошок дарунавира аморфного (USP); стандартный образец дарунавира (MSN Pharmachem Pvt. Ltd., Индия); метанол, класс «ос.ч.», для градиентной ВЭЖХ 99,9 %; ацетонитрил для градиентной ВЭЖХ 99,9 %; уксусная кислота (ледяная) для ВЭЖХ; 0,1 моль/л раствор хлорной кислоты в безводной уксусной кислоте для титрования в неводных средах; нейлоновые шприцевые фильтры с диаметром пор 0,22 мкм. Спектрофотометрическое определение дарунавира проводили, используя спектрофотометр Cary 60 (Agilent Technologies, США) и спектрофотометр UNICO 2800 (United Products & Instruments, Inc., США). Для изготовления стандартных растворов использовали весы аналитические Analytical Balance MS105/A (METTLER TOLEDO, Швейцария), весы аналитические GH-120 (AND, Япония), мерную посуду класса А, градуированные пипетки ISOLAB.
Результаты и обсуждение. Методика была разработана и валидирована по следующим характеристикам: специфичности, линейности, правильности, прецизионности, аналитической области. По результатам исследования основные валидационные характеристики метода соответствуют критериям приемлемости.
Заключение. Была проведена успешная разработка и валидация новой методики количественного определения аморфного дарунавира методом УФ-спектрофотометрии. Методика может быть использована для проведения контроля качества субстанций, действующим веществом которых является аморфный дарунавир, в том числе для внутриаптечного контроля.
Введение. Разработка и регистрация противовирусных препаратов является актуальной задачей. Флавоноиды, в частности лютеолин-7-гликозид (цинарозид, лютеолин-7-О-гликозид), демонстрируют высокую противовирусную активность широкого спектра in vitro, а промышленный регламент получения лютеолин-7-гликозида из листьев ивы остролистной уже разработан в ФГБНУ ВИЛАР. Одной из проблем при внедрении флавоноидов в медицинскую практику является их низкая биодоступность и интенсивная биотрансформация. Существующие публикации приводят противоречивые данные по фармакокинетике лютеолин-7-гликозида, в связи с чем было проведено собственное исследование.
Цель. Разработать методику количественного анализа лютеолин-7-гликозида и его метаболитов в плазме крови и апробировать ее на лабораторных животных.
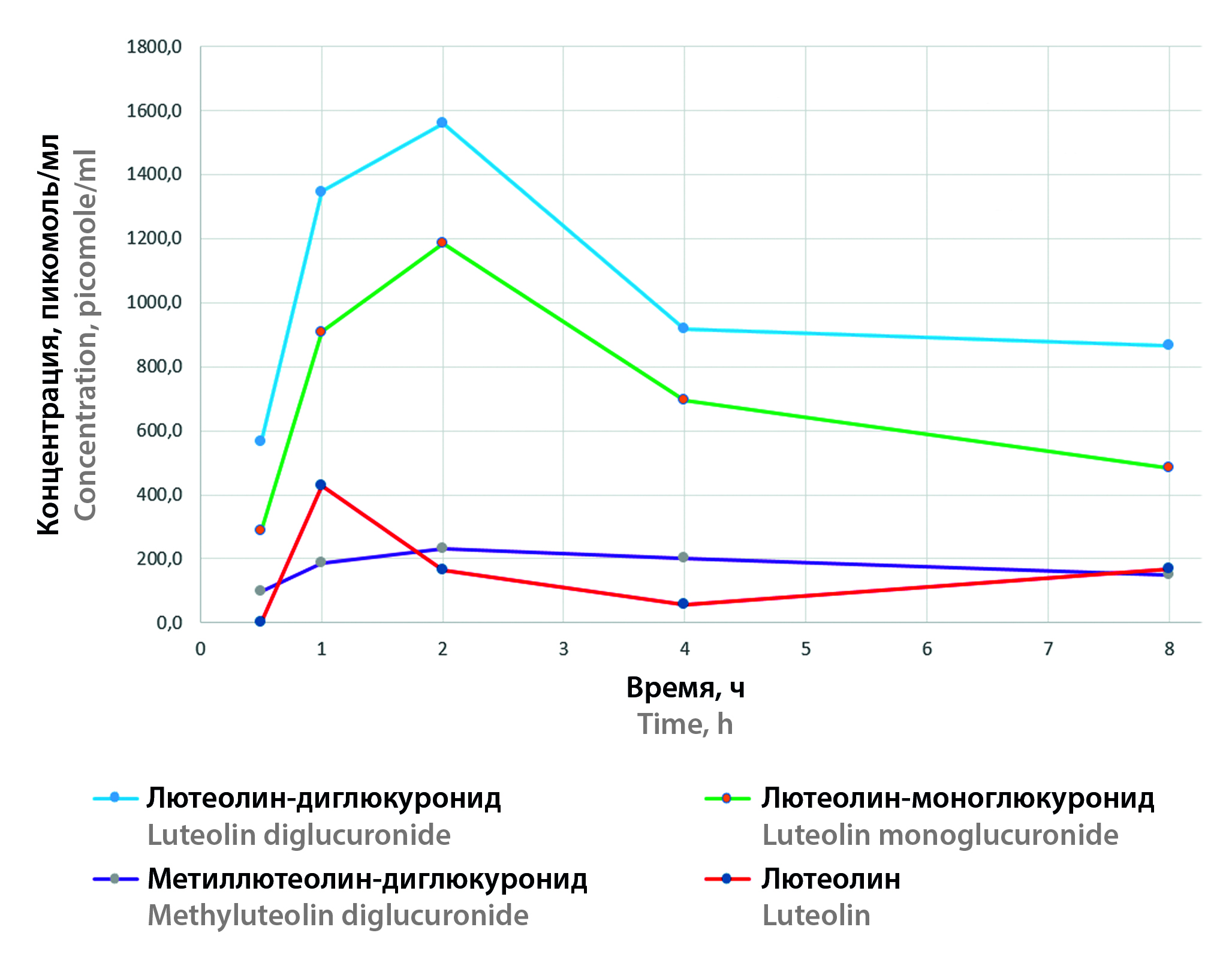
Материалы и методы. Эксперименты на животных проводили согласно требованиям «Руководства по проведению доклинических исследований лекарственных средств». Для разработки методики анализа и дальнейшего уточнения временных интервалов отбора проб крови анализировали временные точки: через 30, 60 минут, 2, 4, 8, 24 часа после введения исследуемого вещества. Пробирки с цитратной кровью лабораторных животных центрифугировали при 2000 оборотов в минуту в течение 10 минут. Плазму помещали в пробирку типа «эппендорф», замораживали и хранили при температуре –20 °С до проведения хроматографического анализа. Пробоподготовку плазмы крови проводили методом осаждения метиловым спиртом, супернатант хроматографически разделяли на колонке Luna® С18 100 Å, 250 × 4,6 мм, 5 мкм, в градиентном режиме в системе «вода – ацетонитрил» и модификатором – 0,2%-й муравьиной кислотой. Метаболиты идентифицировали методом высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектированием. Для этого интерпретировали спектральные характеристики пиков, которые появились на хроматограммах образцов плазмы крови после перорального введения лютеолин-7-гликозида. Концентрацию анализируемых веществ оценивали методом внутреннего стандарта, в качестве которого выступал рутин. Для определения концентрации лютеолина в качестве стандартного образца использовали стандартизированную субстанцию лютеолина, концентрацию остальных метаболитов оценивали в пересчете на лютеолин.
Результаты и обсуждение. Удалось установить, что после перорального введения лабораторным животным лютеолин-7-гликозида в крахмальном клейстере нативный лютеолин-7-гликозид в плазме крови не обнаруживается. Основными метаболитами являлись лютеолин-диглюкуронид и лютеолин-глюкуронид, их максимальные концентрации в плазме почти в три раза выше, чем концентрации лютеолина и метиллютеолина-диглюкуронида. Результаты сопоставлены с данными других исследований.
Заключение. Отсутствие в плазме крови нативного лютеолин-7-гликозида после перорального введения требует пересмотра адекватности выводов, полученных при исследованиях его активности в опытах in vitro. Вместе с тем наличие противовирусной активности in vivo обуславливает необходимость проведения дальнейших исследований для установления реальных механизмов действия данной лекарственной субстанции.
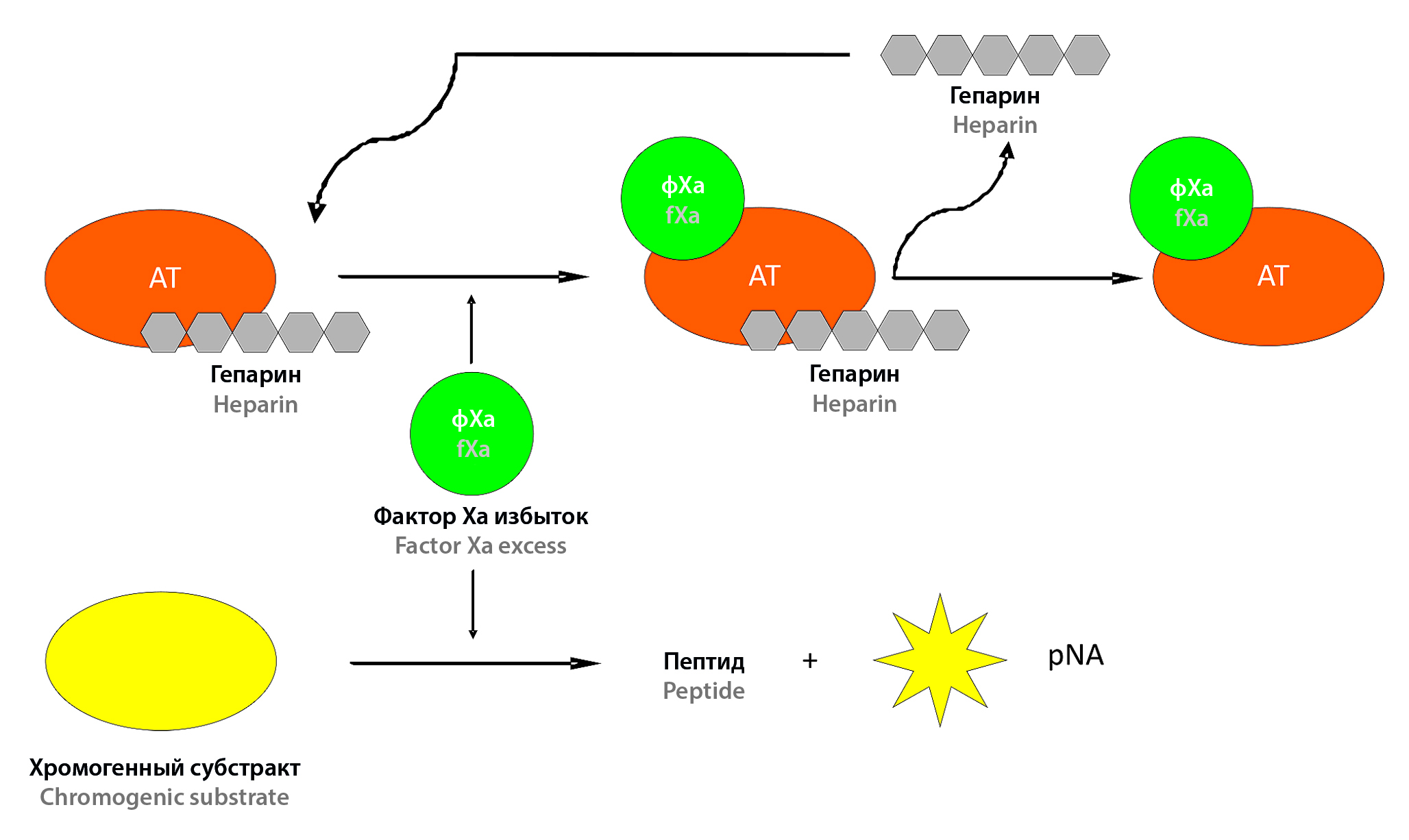
Введение. Представлены данные о широком клиническом применении препаратов низкомолекулярного гепарина (НМГ), обуславливающем необходимость эффективного и качественного производства соответствующих фармацевтических средств. Выявленная вариабельность антикоагулянтной активности препаратов НМГ предполагает обеспечение производства валидированными методами контроля и аттестации. Цель работы заключается в изучении возможности контроля антикоагулянтной активности и оценки функциональной совместимости препаратов НМГ при применении отечественных тест-систем.
Текст. Представлены данные об аналитических характеристиках хромогенного и коагулометрических методов, выполняемых с помощью отечественных тест-систем. Показано соответствие их показателей существующим международным требованиям. Доказана возможность указанных методов правильно и воспроизводимо сертифицировать препараты НМГ. Полученные в рамках исследований биоэквивалентности результаты свидетельствуют о возможности коагулологических методик оценивать функциональную совместимость изучаемых препаратов НМГ.
Заключение. Представленные данные доказывают целесообразность проведения контроля производства и аттестации препаратов НМГ отечественными тест-системами.
ДОКЛИНИЧЕСКИЕ И КЛИНИЧЕСКИЕ ИССЛЕДОВАНИЯ
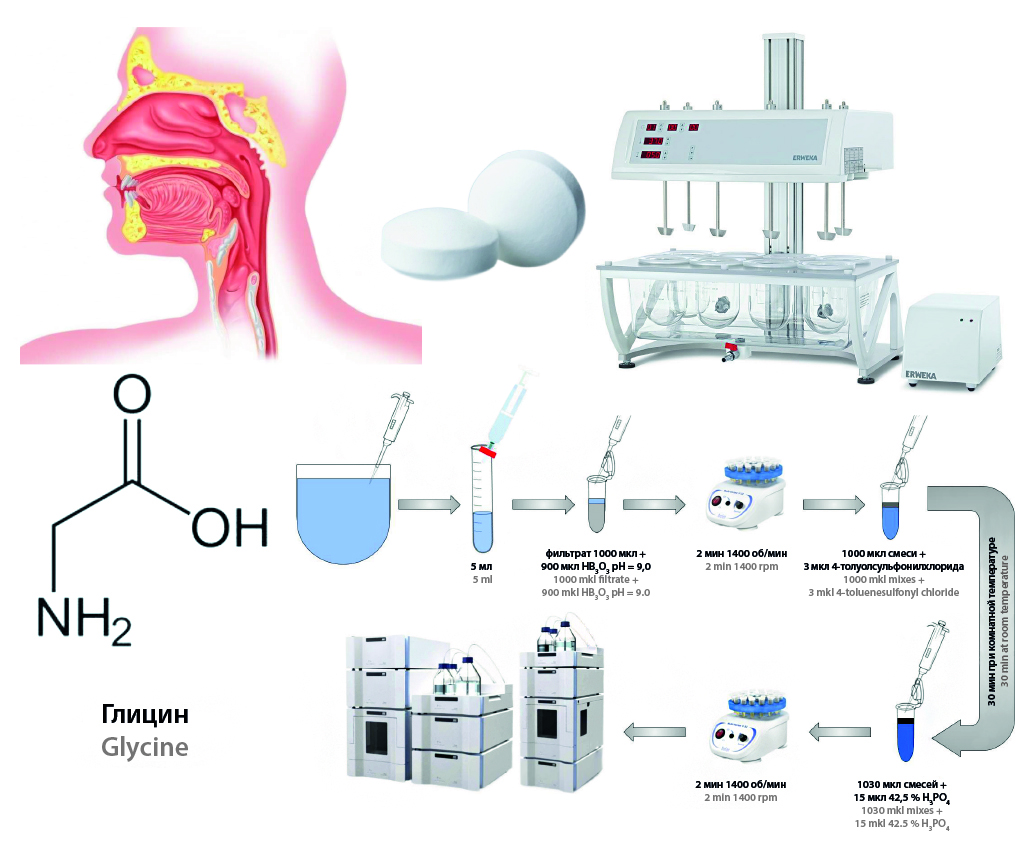
Введение. Биорелевантные среды растворения воссоздают состав содержимого желудочно-кишечного тракта. Они используются в качестве среды растворения при оценке профилей растворения различных лекарственных форм. Имитированные биологические жидкости позволяют прогнозировать результаты испытаний in vivo. Разработка состава имитированной слюнной жидкости позволяет оценить свойства лекарственного препарата в физиологически релевантных условиях.
Цель. Оценка высвобождения лекарственного препарата «глицин, таблетки подъязычные, 100 мг» отечественного производства в среду растворения Simulated Saliva 5, pH 6,8.
Материалы и методы. Для анализа использовались препараты: «Глицин, таблетки подъязычные, 100 мг» отечественного производства с действующим сроком годности. Тест сравнительной кинетики растворения проводили на приборе для теста «Растворение» DT 6 (ERWEKA GmbH, Германия). Хроматографическое разделение и детектирование проводили на высокоэффективном жидкостном хроматографе Waters W1525 Binary HPLC Pump (Waters Corporation, США), оснащенном термостатом колонок и образцов, дегазатором, автосамплером и ультрафиолетовым детектором Waters 2487 Dual Absorbance Detector (Waters Corporation, США). Детектирование проводилось при длине волны 254 ± 2 нм после дериватизации молекулы глицина 4-толуолсульфонилхлоридом. Использовали колонку Grace Platinum C18-EPS, 4,6 × 250 мм, 5 мкм (Grace, США) и предколонку Grace Platinum C18-EPS, 4,6 × 250 мм, 5 мкм (Grace, США). Для исследования использовалось следующее программное обеспечение: валидированная автоматическая таблица Microsoft Excel для расчета значений высвобождения глицина.
Результаты и обсуждение. Разработана и валидирована методика количественного определения глицина в рамках ТСКР в среде воды очищенной и среде, имитирующей слюну человека, Simulated Saliva 5, pH 6,8. Подтвержденный аналитический диапазон методики составил 10–110 % от номинальной концентрации лекарственной формы в объеме среды 300 мл. Разработанная аналитическая методика была апробирована в ходе проведения биопредиктивного in vitro теста препаратов глицина. При проведении исследования в среде Simulated Saliva для лекарственных препаратов были получены более дискриминативные данные по сравнению со средой растворения «вода очищенная», что выражалось в разной скорости растворения, кривизне наклона профиля растворения и времени выхода на плато.
Заключение. Разработана и валидирована методика количественного определения для проведения биопредиктивных тестов таблеток «Глицин, таблетки подъязычные, 100 мг». Аналитический диапазон методики составил 10–110 % от номинальной концентрации лекарственной формы в объеме среды 300 мл. Результаты проведения теста в среде искусственной слюны обладали большей дискриминативностью в сравнении с водой очищенной и позволили обнаружить различия в полноте высвобождения лекарственных препаратов, времени достижения плато и угла наклона кривой профиля растворения.
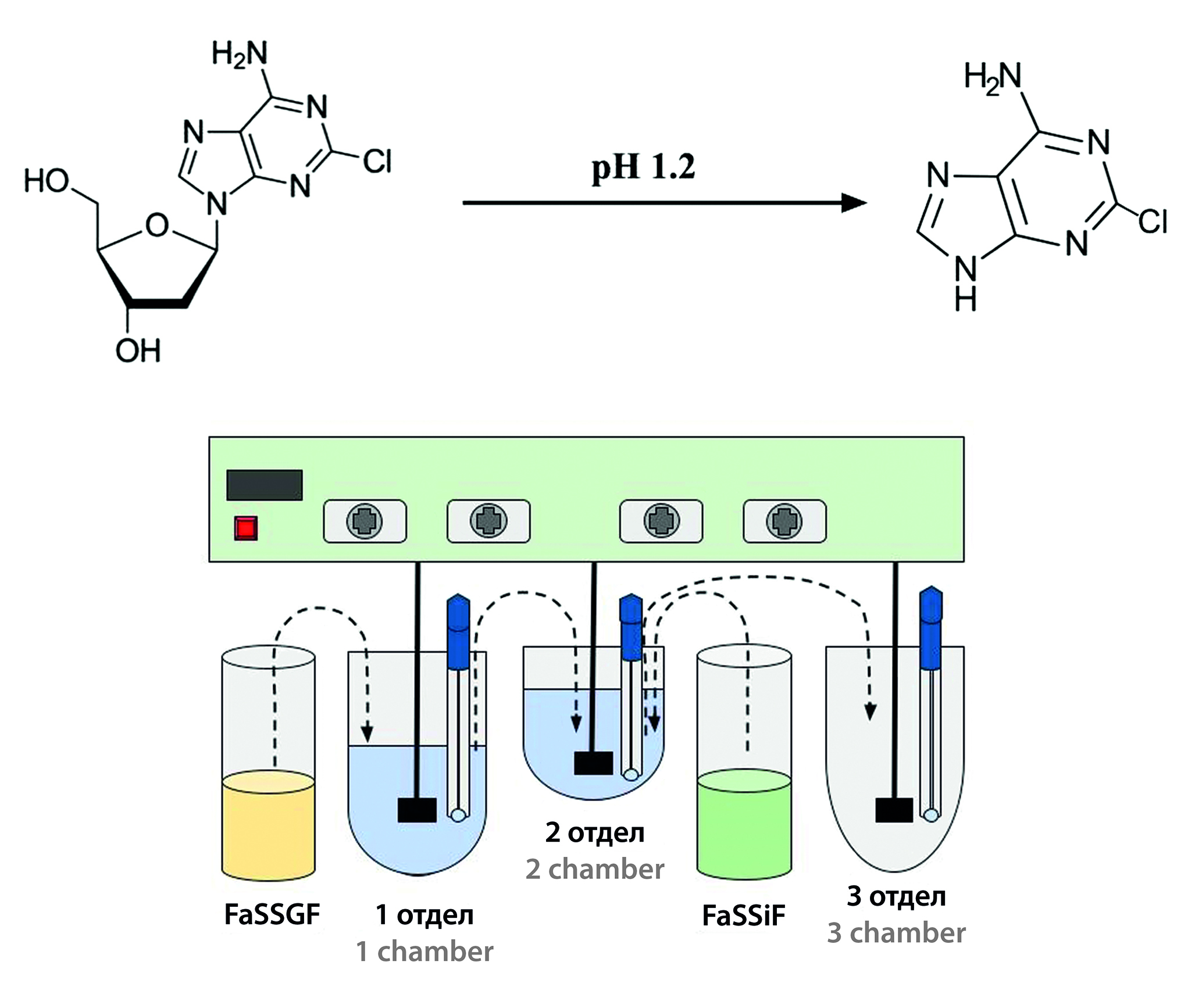
Введение. Внедрение аппаратов – аналогов GIS (далее – Gastro-intestinal simulator) является одним из актуальных путей развития in-vitro-оценки качества твердых лекарственных форм. Испытания на приборе для физиологически релевантных тестов (далее – ФРТ) позволяют предсказать фармакокинетические профили за счет более релевантных условий, среди которых использование биорелевантных сред растворения, физиологичные объемы отделов ЖКТ, а также транзит между ними.
Цель. Провести исследование таблеток кладрибина на физиологически релевантном тестере с целью предсказания поведения препарата в ЖКТ человека.
Материалы и методы. Объектами исследования являются «Мавенклад®, таблетки, 10 мг» (серия 2200754, срок годности до 04.2025, NERPHARMA, S.r.L., Италия) и «Кладрибин, таблетки, 10 мг» отечественного производства с действующим сроком годности. Во время исследования использовались реактивы, необходимые для приготовления биорелевантных сред растворения и проведения количественного определения методом ВЭЖХ. Физиологически релевантный тест проводили на аппарате собственного производства, состоящем из тестера растворения DT-6 (ERWEKA GmbH, Германия), водяной бани, оснащенной нагревательным элементом Thermomix WB-4 (B. Braun, Германия), насосов перистальтических (Kamoer, Китай). Количественное содержание высвободившегося кладрибина оценивали на высокоэффективном жидкостном хроматографе «Хроматэк-Кристалл ВЭЖХ 2014» (ЗАО СКБ «Хроматэк», Россия) по валидированной методике при длине волны 252 нм, время анализа – 7 мин, колонка – Grace HPLC Column Platinum C18-EPS, 250 × 4.6 мм, 5 мм (Grace, США), температура – 35 °С, режим элюирования – изократический (А : В 80 : 20), подвижная фаза А – 0,1%-й раствор H3PO4, фаза В – ацетонитрил.
Результаты и обсуждение. Были получены профили, позволяющие оценить динамику и степень высвобождения исследуемых ЛС в различных отделах ЖКТ человека. Несмотря на ожидаемую деградацию кладрибина в кислой среде (рН 1,2), в физиологически релевантных условиях препарат достиг третьего отдела (модель тонкого кишечника) без деградации. Наблюдалось полное высвобождение кладрибина из лекарственной формы для тестового и референтного лекарственных препаратов. Также в дальнейшем, исходя из полученных данных, можно предсказать фармакокинетические профили при помощи подходов физиологически обоснованного фармакокинетического моделирования.
Заключение. Проведено исследование ФРТ для препаратов «Мавенклад®, таблетки, 10 мг» и «Кладрибин, таблетки, 10 мг». Количественное определение проводилось методом ВЭЖХ-УФ. По результатам испытания было отмечено полное высвобождение обоих препаратов и достижение отдела, имитирующего кишечник, что указывает на отсутствие деградации кладрибина в отделе, имитирующем желудок.
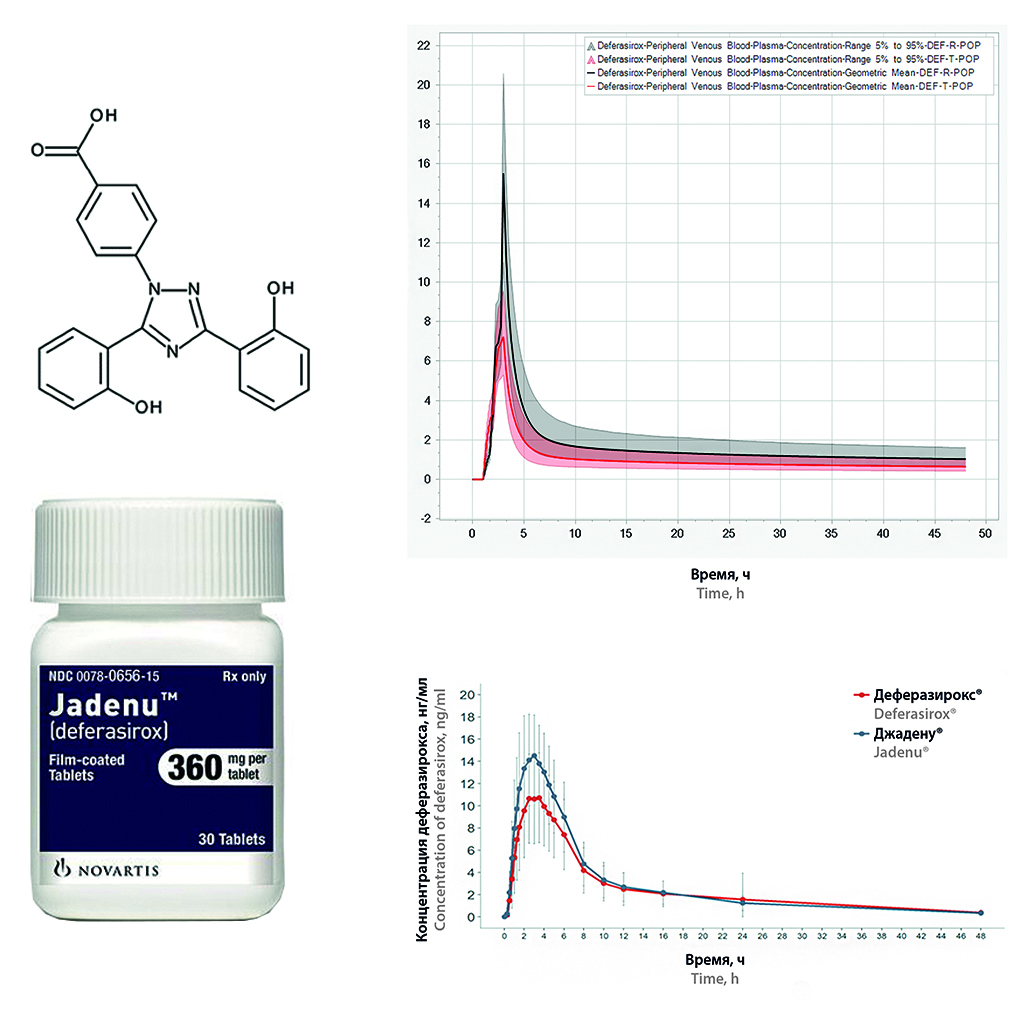
Введение. Деферазирокс является комплексообразующим лекарственным средством и относится ко II классу по биофармацевтической классификационной системе (БКС), обладает кислотными свойствами и относится к подклассу «а» (acid). Данный класс характеризуется высокой проницаемостью и низкой растворимостью, которая лимитирует всасывание действующего вещества в кровь. Вследствие этого разработка препаратов с действующим веществом, которое можно отнести к данному классу БКС, является сложной задачей, а для воспроизведенных препаратов еще и сопряжена с высоким риском получения недоказанной эквивалентности при проведении клинических исследований. Для минимизации вышеописанных рисков был проведен физиологически релевантный тест с дальнейшей обработкой данных и построением предполагаемых фармакокинетических профилей.
Цель. Целью исследования является проведение физиологически релевантного теста (ФРТ) для предсказания по данным in vitro фармакокинетических профилей и сопоставление с данными in vivo в рамках исследования биоэквивалентности препарата деферазирокс.
Материалы и методы. Объектами исследования являются «Деферазирокс, таблетки, покрытые пленочной оболочкой, 360 мг» отечественного производителя и «Джадену®, таблетки, покрытые пленочной оболочкой, 360 мг» (серия WTN22, срок годности до 10.2023, Novartis Pharma Stein AG, Швейцария). Физиологически релевантный тест проводили на приборе СК ФРТ-6 (ООО «Сайнтифик Комплайнс», Россия). Количественный анализ проводили методом ВЭЖХ-УФ. Фармакокинетические профили были смоделированы в программе PK-Sim® (Systems Biology Software Suite 11.2, Bayer Technology Services GmbH, Германия) на основании данных, полученных в рамках проведения физиологически релевантного теста.
Результаты и обсуждение. Был проведен физиологически релевантный тест для лекарственных препаратов деферазирокса, получены профили высвобождения, которые легли в основу физиологически обоснованной фармакокинетической модели совместно с данными о физико-химических свойствах изучаемого соединения и литературными данными о фармакокинетике деферазирокса. Полученные в рамках симуляции на виртуальной популяции фармакокинетические профили были сопоставлены с данными, полученными при проведении клинических испытаний.
Заключение. Проведен физиологически релевантный тест для препарата деферазирокс, количественное определение в образцах проводили методом ВЭЖХ-УФ. В результате проведения теста были получены данные, позволившие спрогнозировать фармакокинетические профили, которые отражают те же различия, что наблюдались в профилях тестового и референтного препарата при проведении исследования сравнительной фармакокинетики и биоэквивалентности препаратов деферазирокса.
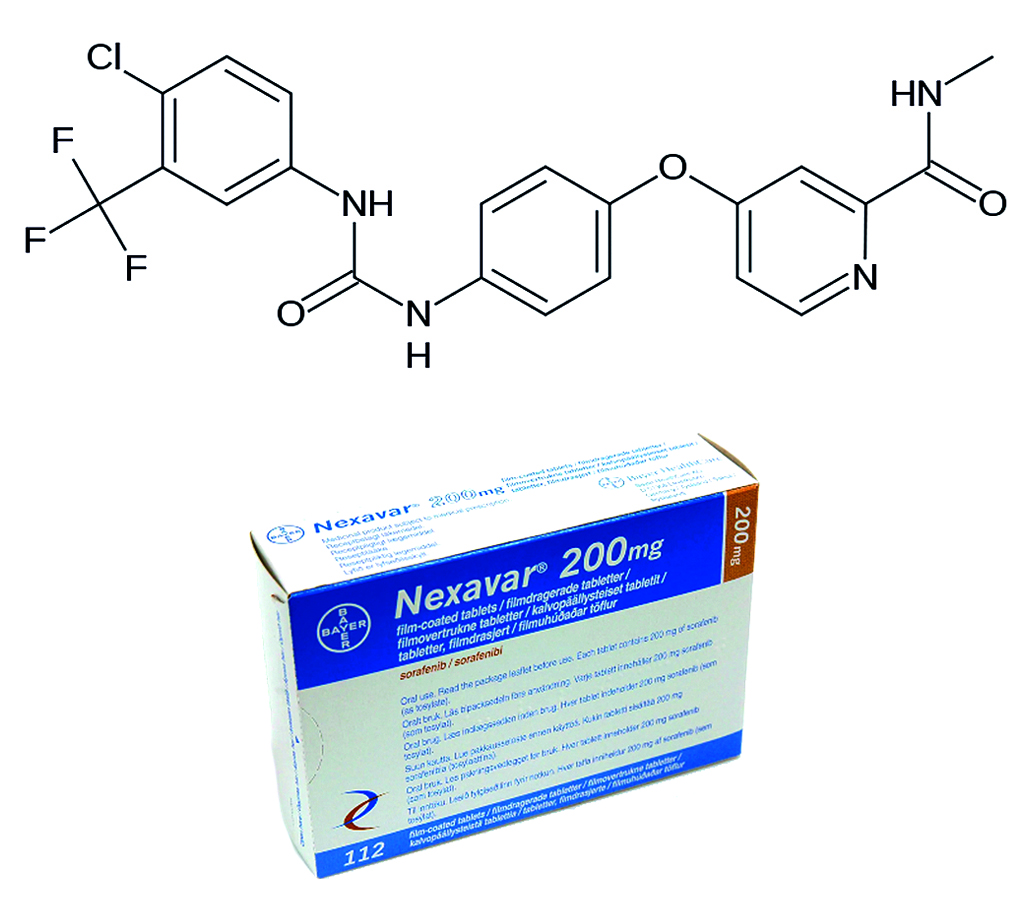
Введение. Сорафениб – противоопухолевое лекарственное средство, относящееся к классу IIс по биофармацевтической классификационной системе (БКС) за счет наличия и кислотных, и основных свойств. Кроме низкой растворимости, сорафениб характеризуется высокой вариабельностью при проведении клинических исследований, в частности исследований биоэквивалентности (БЭ). Для целей выбора серий, которые могут быть рекомендованы при проведении исследований БЭ, в настоящее время широко применяется тест кинетики растворения, однако результатов данного теста не всегда достаточно и проведение дополнительных тестов, например физиологически релевантного теста, является целесообразным. Для минимизации рисков получения неэквивалентных результатов при проведении исследования БЭ был проведен физиологически релевантный тест (ФРТ) с дальнейшей обработкой данных и интерпретацией результатов физиологически обоснованного фармакокинетического моделирования (ФОФМ).
Цель. Целью исследования является проведение физиологически релевантного теста (ФРТ) для целей выбора с применением ФОФМ (физиологически обоснованное фармакокинетическое моделирование, physiologically based pharmacokinetic model, PBPK) серии-кандидата для последующего исследования БЭ препаратов сорафениба.
Материалы и методы. Объектами исследования являются «Нексавар®, таблетки, покрытые пленочной оболочкой, 200 мг» (одна серия) (Bayer AG, Германия) и «Сорафениб, таблетки, покрытые пленочной оболочкой, 200 мг» (две серии) (Россия). Физиологически релевантный тест проводили на приборе СК ФРТ-6 (ООО «Сайнтифик Комплайнс», Россия). Количественный анализ проводили методом ВЭЖХ-УФ на приборе «Хроматэк-Кристалл ВЭЖХ 2014» (ЗАО СКБ «Хроматэк», Россия). Моделирование профилей «плазма – концентрация» проводилось с помощью программного обеспечения PK-Sim® (Systems Biology Software Suite 11.2, Bayer Technology Services GmbH, Германия).
Результаты и обсуждение. В рамках выполнения исследований была разработана и валидирована методика количественного определения сорафениба, разработана методика пробоподготовки и методика проведения ФРТ для сорафениба как представителя подкласса IIс БКС. По результатам исследования получены профили высвобождения, которые были использованы для целей выбора серии кандидата для проведения исследования БЭ. Выбор серий производился на основании ФОФМ-анализа на виртуальной популяции, состоящей из 36 здоровых добровольцев с активированной энтеропатической циркуляцией, характерной для сорафениба.
Заключение. Проведен ФРТ для препарата сорафениб. Количественное определение проводилось методом ВЭЖХ-УФ по разработанной и валидированной методике. В результате проведения теста были получены данные, подвергнутые ФОФМ-анализу. Было показано, что исследованные серии имеют высокие риски получения результатов с недоказанной эквивалентностью при проведении клинического исследования.
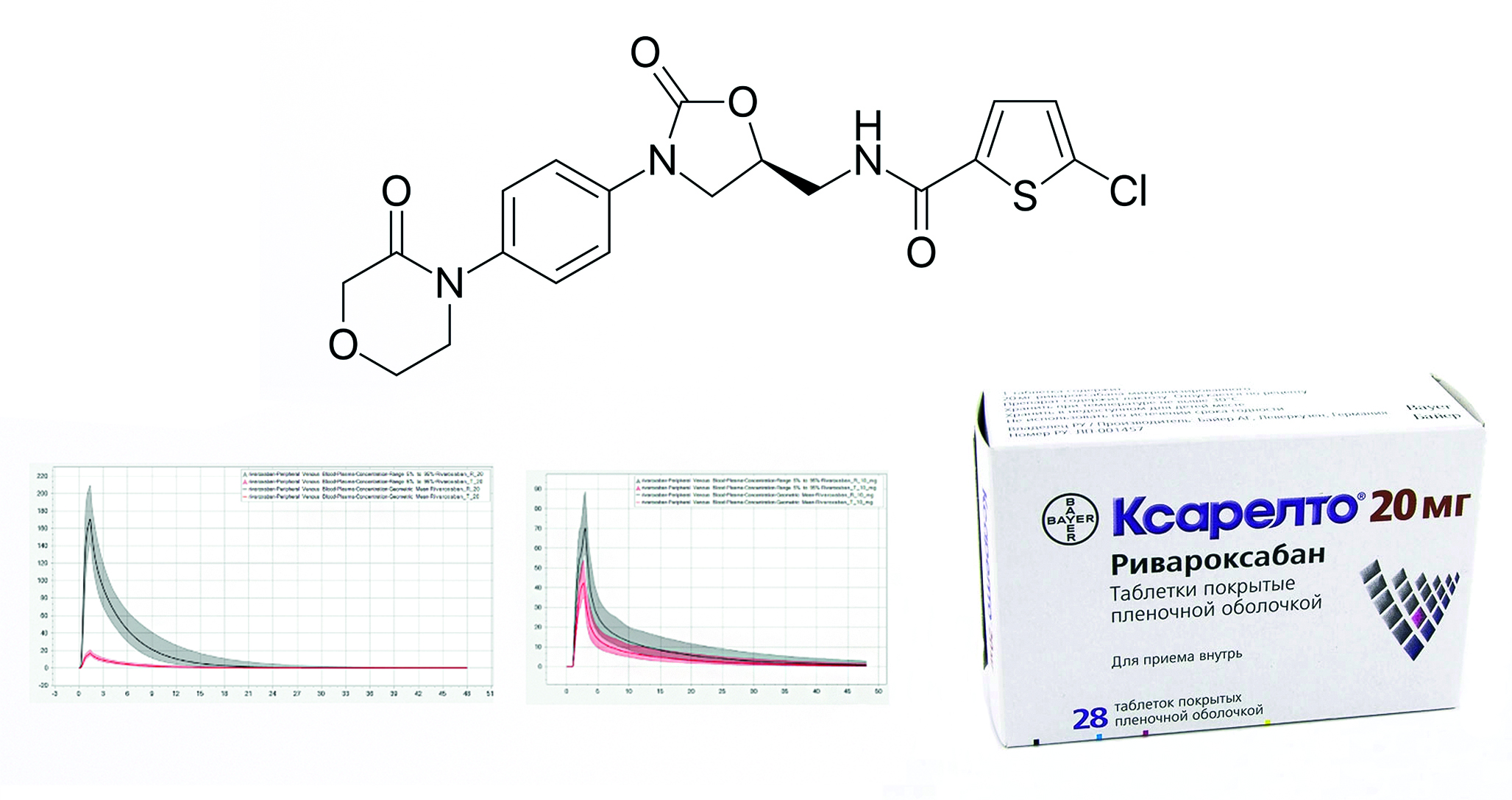
Введение. Важнейшим этапом фармацевтической разработки воспроизведенного лекарственного средства (ЛС) является клиническое исследование с участием человека – исследование биоэквивалентности. Учитывая важность нахождения препаратов ривароксабана в перечне жизненно необходимых и важнейших лекарственных препаратов (ЖНВЛП), в рамках обеспечения технологического суверенитета требуется использование научных робастных и эффективных методов определения качества готовой лекарственной формы.
Цель. Провести исследование таблеток ривароксабана на физиологически релевантном тестере с целью предсказания фармакокинетических профилей.
Материалы и методы. Объектами исследования являются «Ксарелто®, таблетки, покрытые пленочной оболочкой, 10 мг» (серия BXJS871, срок годности до 31.10.2024, Bayer AG, Германия), «Ксарелто®, таблетки, покрытые пленочной оболочкой, 20 мг» (серия BXKDF32, срок годности до 17.05.2026, Bayer AG, Германия), «Ривароксабан, таблетки, покрытые пленочной оболочкой, 10 мг» и «Ривароксабан, таблетки, покрытые пленочной оболочкой, 20 мг» отечественного производства, с действующими сроками годности. Во время исследования использовались реактивы, необходимые для приготовления сред растворения и проведения количественного определения. Физиологически релевантный тест проводили на приборе СК ФРТ-6 (ООО «Сайнтифик Комплайнс», Россия). Определение количественного содержания высвободившегося ривароксабана в рамках теста сравнительной кинетики растворения (ТСКР) в среде 0,1%-го раствора натрия лаурилсульфата в фосфатном буферном растворе с рН 6,5 проводилось на спекторофотометре СФ-2000 (ООО «ОКБ Спектр», Россия). Количественное содержание высвободившегося ривароксабана в рамках ТСКР в биорелевантных средах растворения (БРС) и физиологически релевантного теста (ФРТ) оценивали на высокоэффективном жидкостном хроматографе «Хроматэк-Кристалл ВЭЖХ 2014» (ЗАО СКБ «Хроматэк», Россия). Фармакокинетические профили были смоделированы в программе PK-Sim® (Systems Biology Software Suite 11.2, Bayer Technology Services GmbH, Германия) на основании данных, полученных в рамках проведения ФРТ. Клиническое исследование таблеток ривароксабана представляло собой проспективное открытое рандомизированное перекрестное в двух этапах сравнительное исследование в двух группах добровольцев с однократным приемом препаратов натощак. В исследовании были рандомизированы 30 здоровых добровольцев мужского пола в возрасте 18–45 лет.
Результаты и обсуждение. Был проведен комплекс испытаний in vitro, получены профили, позволяющие оценить динамику и степень высвобождения исследуемых ЛС в различных отделах ЖКТ человека. Осуществлено сравнение последовательной и гибридной схем проведения ФРТ. По результатам проведения ФРТ по разным схемам были предсказаны фармакокинетические профили для пары препаратов и рассчитана ошибка прогнозирования.
Заключение. Проведен комплекс научных испытаний in vitro для препаратов «Ксарелто®, таблетки, покрытые пленочной оболочкой, 10 мг и 20 мг», «Ривароксабан, таблетки, покрытые пленочной оболочкой, 10 мг и 20 мг». В рамках сравнения данных, полученных при проведении клинического исследования и при моделировании наименьшая ошибка прогнозирования была отмечена при выполнении ФРТ по гибридной схеме.
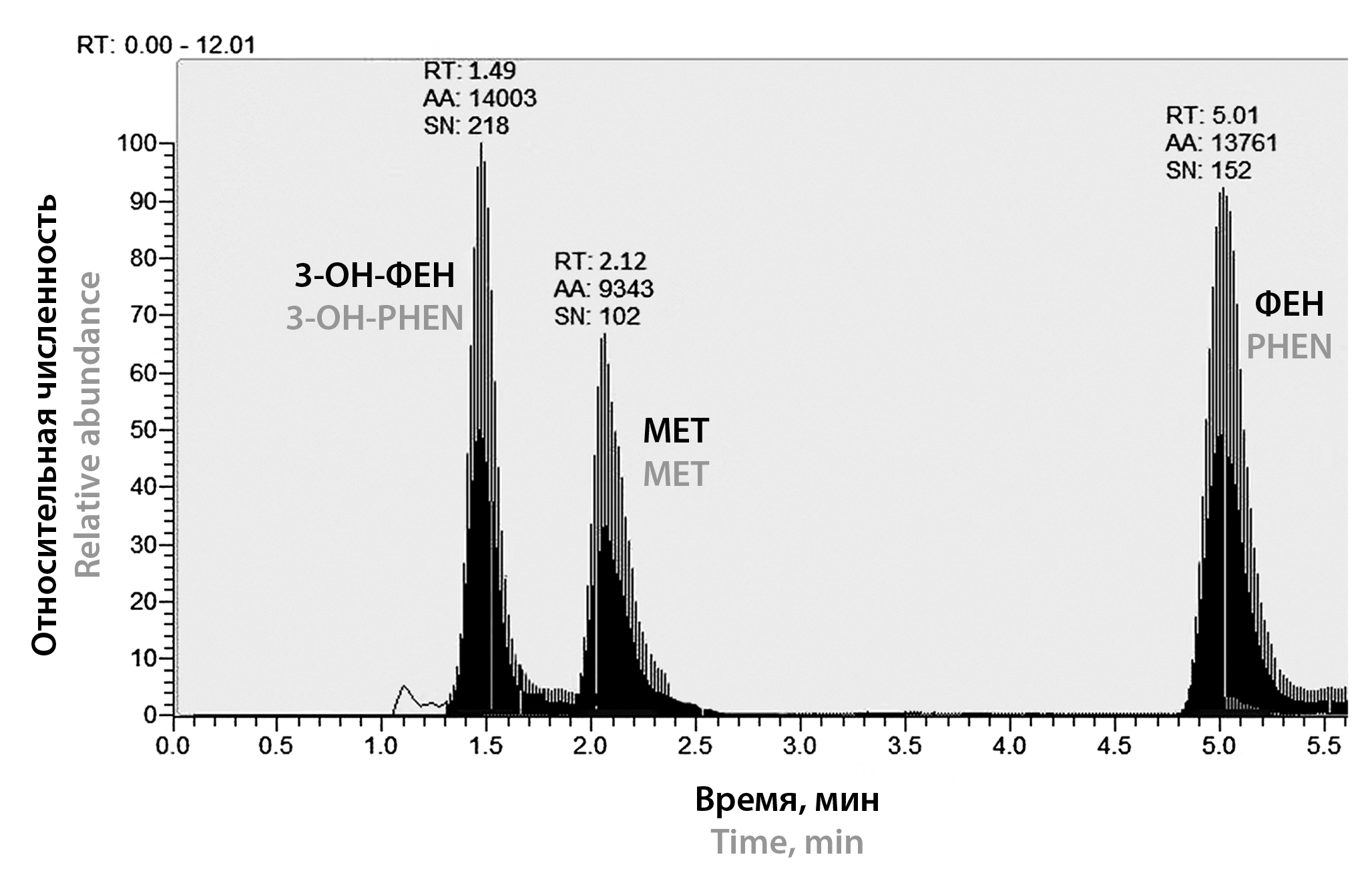
Введение. Наличие активного метаболита 3-гидроксифеназепама (3-ОН-ФЕН), широкая межиндивидуальная вариабельность активной фракции феназепама (ФЕН) в крови, а также его терапевтического эффекта определяют актуальность терапевтического лекарственного мониторинга (ТЛМ). Для этого исследователь должен располагать экспрессной методикой с широким аналитическим диапазоном, низким пределом количественного определения (НПКО), робастной при разных методах пробоподготовки.
Цель. Разработка и валидация методик количественного определения ФЕН и 3-гидроксифеназепама для измерения содержания этих аналитов в плазме крови человека при разных методах экстракции.
Материалы и методы. Определение ФЕН и 3-ОН-ФЕН в плазме крови человека проводили методом высокоэффективной жидкостной хроматографии с тандемной масс-спектрометрией (ВЭЖХ-МС/МС). Для пробоподготовки использовали твердофазную экстракцию (SPE, от англ. «solid phase extraction») и жидкостную экстракцию с поддержкой (SLE, от англ. «supported liquid extraction»), иначе называемую жидкостно-жидкостной экстракцией на твердой фазе. Внутренний стандарт (ВС) − метопролол. Подвижная фаза: 0,2%-й водный раствор муравьиной кислоты (элюент А) и 100%-й ацетонитрил (элюент B); режим: градиентный. Колонка: Hypersil GOLD® C18, 50 × 2,1 мм, 3,5 мкм.
Результаты и обсуждение. Разработаны две методики количественного определения ФЕН и 3-ОН-ФЕН в плазме крови человека при разных методах пробоподготовки: SPE и SLE. Подобраны условия хроматографического разделения и масс-спектрометрической детекции изучаемых аналитов. Для обеих методик были определены следующие валидационные характеристики: селективность, калибровочная кривая, точность, прецизионность, степень экстракции, НПКО, эффект переноса, фактор матрицы, стабильность рабочих растворов и аналита в матрице.
Заключение. Результаты валидации разработанных методик соответствуют установленным критериям, что позволяет их использовать для количественного определения ФЕН и 3-ОН-ФЕН в плазме крови человека. Широкий аналитический диапазон обеих методик – 1–1000,00 нг/мл – позволяет применять их для проведения исследований фармакокинетики и биоэквивалентности, а также в токсикологии.
Введение. Применение доксорубицина в клинической практике сопровождается кумулятивным и дозозависимым токсическим воздействием на кардиомиоциты, приводящим к увеличению риска смертности среди пациентов с онкологическими заболеваниями и, как следствие, возникновению ограничений в отношении его применения.
Текст. Опасной нежелательной реакцией доксорубицина является кардиомиопатия, приводящая к застойной сердечной недостаточности. В основе кардиотоксичности лежат как минимум несколько патофизиологических механизмов (детальнее описанных в первой части обзора), приводящих к повреждению кардиомиоцитов в результате окислительного стресса с образованием свободных радикалов, нарушения функции митохондрий, аутофагии, высвобождения оксида азота и медиаторов воспаления, а также изменения экспрессии генов и белков, что приводит к апоптозу. В текущей (второй) части обзора представлена подробная информация о современном понимании патофизиологических механизмов, лежащих в основе уже описанной кардиотоксичности, и влияния доксорубицина на другие клетки сердца. Использование кардиопротективных стратегий позволит снизить выраженность и вероятность развития кардиотоксичности. В данной статье описаны стратегии, основанные на снижении максимальной кумулятивной дозы, изменении характера введения доксорубицина, использовании пегилированных липосомальных форм и кардиопротекстивных средств, а также физических нагрузок.
Заключение. Несмотря на огромное количество научных работ, посвященных различным аспектам кардиотоксичности доксорубицина, ее профилактики и лечения, данный вопрос требует более тщательного изучения и выработки более совершенных методов ранней диагностики, профилактики и более эффективной терапии этого осложнения.
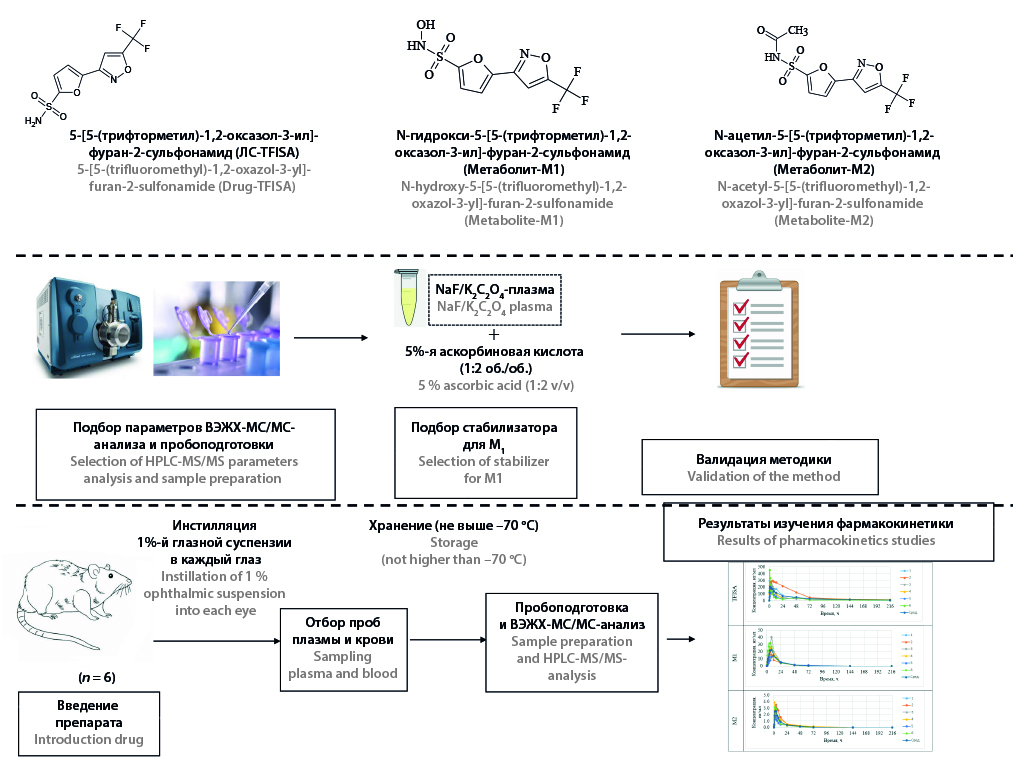
Введение. Изучение системной экспозиции нового оригинального лекарственного препарата является обязательным этапом его доклинического исследования. 5-[5-(трифторметил)-1,2-оксазол-3-ил]-фуран-2-сульфонамид является новым селективным ингибитором карбоангидразы II для лечения открытоугольной глаукомы. Методики количественного определения данного соединения и его N-гидрокси- и N-ацетилметаболитов в плазме лабораторных животных ранее разработаны не были.
Цель. Разработка и валидация методики количественного определения 5-[5-(трифторметил)-1,2-оксазол-3-ил]-фуран-2-сульфонамида и его метаболитов N-гидрокси-5-[5-(трифторметил)-1,2-оксазол-3-ил]-фуран-2-сульфонамида (М1) и N-ацетил-5-[5-(трифторметил)-1,2-оксазол-3-ил]-фуран-2-сульфонамида (М2) в плазме крови крысы и кролика методом ВЭЖХ-МС/МС.
Материалы и методы. Для пробоподготовки применялось осаждение белков метанолом. В качестве внутреннего стандарта использован 5-[2-(морфолин-4-карбонил)-1,3-оксазол-5-ил]-тиофен-2-сульфонамид. К пробам плазмы добавлялся 5%-й водный раствор аскорбиновой кислоты в объемном соотношении 1 : 2 для предотвращения разложения N-гидрокси-5-[5-(трифторметил)-1,2-оксазол-3-ил]-фуран-2-сульфонамида. В качестве антикоагулянта выбрана комбинация натрия фторида и калия оксалата. Хроматографическое разделение проводили на колонке Zorbax Eclipse Plus C18 (150 × 3,0 мм, 3,5 мкм) с предколонкой Zorbax Eclipse Plus C18 (12,5 × 2,1 мм, 5,0 мкм) с применением подвижной фазы на основе 0,1%-го водного раствора муравьиной кислоты и метанола. Масс-спектрометрическое детектирование осуществляли в режиме MRM с использованием электрораспылительной ионизации в отрицательной полярности. Апробацию методики проводили во время фармакокинетического исследования 1%-й глазной суспензии 5-[5-(трифторметил)-1,2-оксазол-3-ил]-фуран-2-сульфонамида на 6 крысах линии Wistar. Образцы крови отбирали до введения, а также спустя 30 мин, 1 ч, 1ч 30 мин, 2 ч, 3 ч, 4 ч, 6 ч, 8 ч, 12 ч, 24 ч, 48 ч, 72 ч,144 ч, 216 ч после введения. Некомпартментный подход использовался для расчета фармакокинетических параметров.
Результаты и обсуждение. Разработанная методика прошла валидацию по показателям «селективность», «калибровочная кривая», «правильность и прецизионность», «эффект матрицы», «эффект разведения», «эффект переноса из предыдущей пробы», «воспроизводимость при повторном введении серии», «стабильность». Аналитический диапазон определения в плазме 5-[5-(трифторметил)-1,2-оксазол-3-ил]-фуран-2-сульфонамида составил 10–4000 нг/мл, М1 – 1,0–400,0 нг/мл, М2 – 0,1–40,0 нг/мл. Выбранная комбинация антикоагулянта и раствора стабилизатора позволяет хранить образцы плазмы в течение 28 дней в морозильной камере.
Заключение. Разработанная методика в ходе проведённой валидации подтвердила свою пригодность для количественного определения 5-[5-(трифторметил)-1,2-оксазол-3-ил]-фуран-2-сульфонамида и его метаболитов в плазме крови лабораторных животных. Данная методика успешно использована при исследовании фармакокинетики 1%-й глазной суспензии изучаемого препарата.

ISSN 2658-5049 (Online)



































_2.jpg")


